 腾讯登录
腾讯登录细胞异质性研究策略解析
| 导读 | 研究细胞异质性,是一个单细胞层面的范畴。单细胞间的异质性存在于DNA、RNA、蛋白等各个层面。在此,我们将结合具体案例,介绍从单细胞转录组着手,通过高通量数据分析方法揭示单细胞异质性的研究思路。 |
细胞的异质性 (heterogeneity) 是一个普遍存在的生物学现象。多细胞生物个体由多种形态功能不同的细胞组成。多种类型细胞有序地结合在一起,形成了组织和器官。在疾病发生的情况下,异常的细胞常常藏匿于正常细胞之中。肿瘤组织也具有很强的细胞异质性,其中决定肿瘤发展方向的细胞可能只占整个肿瘤组织的一小部分。而且近年研究表明,即使看起来相同的细胞,也可能存在显著的异质性。
目前,通过高通量的分子技术平台,对一定数量的单细胞的进行基因表达水平的检测(RNA或蛋白),是揭示细胞异质性最有效的途径之一。
法国国家科研中心分子细胞及遗传学研究所I Davidson团队通过对黑色素瘤单细胞进行基因表达水平的研究,发现了黑色素瘤两个亚类细胞群(增殖型细胞和侵染(invasive)型细胞)之间异质性产生的分子基础,并为临床肿瘤检测提供了全新的思路和线索(ref. 1)。
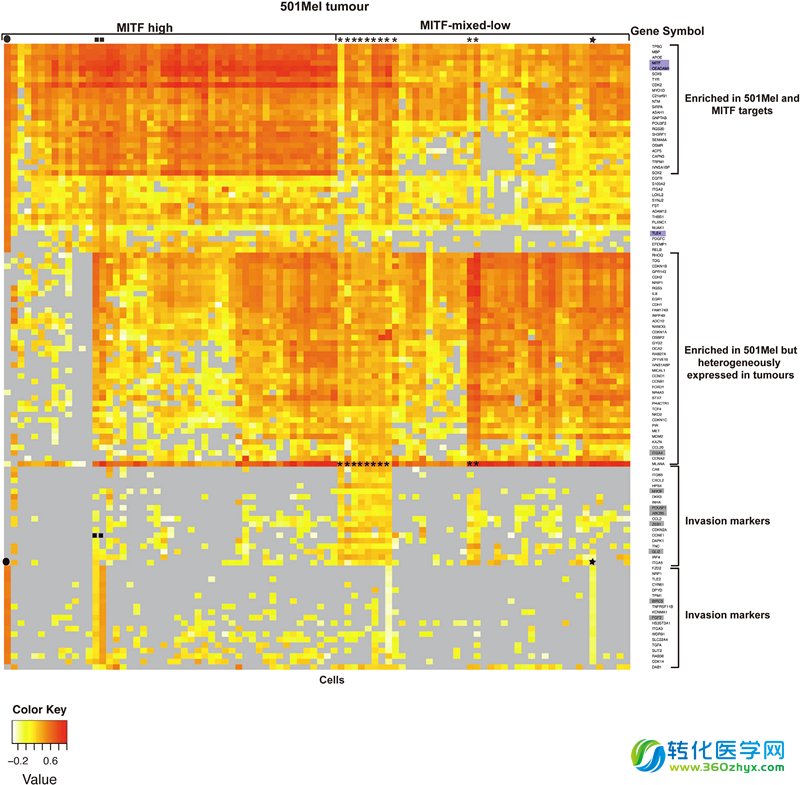
肿瘤并不是均一的细胞团,肿瘤组织的不同细胞具有不同的侵染增殖能力及成瘤潜能。传统的抗肿瘤药物对增殖速度快的肿瘤细胞清除效果较好,但对已发生转变并获得耐药性的细胞则效果十分有限。I Davidson研究团队将一株增殖速度快但侵染能力差的黑色素瘤细胞注入小鼠皮下培养成瘤,待12周后将实体瘤取出,利用Fluidigm C1全自动单细胞制备系统获得该肿瘤单细胞的cDNA样本,并利用Biomark高通量qPCR仪对92个单细胞的113个基因进行了检测。引人注目的是,他们在少量肿瘤单细胞中检测出了侵染(invasive)相关基因(ZEB1,GLI2,MYOF)和耐药基因(ABCBS)的表达(图1)。
“我们利用单细胞的检测手段,发现在原本低侵染性的黑色素瘤细胞中,一小部分已经转变成了高侵染性的肿瘤细胞,并且还具备了耐药性。”作者说道。“相比起传统的活检手段,针对肿瘤单细胞进行分析,能更好的揭示肿瘤异质性,挖掘新的肿瘤标记,揭示肿瘤形成的过程。”
美国Dana-Farber肿瘤研究中心的Guo-Cheng Yuan团队综合运用多种高通量数据的分析手段,首次发现白血病细胞可以分为两个亚群,而且白血病肿瘤干细胞可能匿藏于其中一个亚群中(ref. 2)。
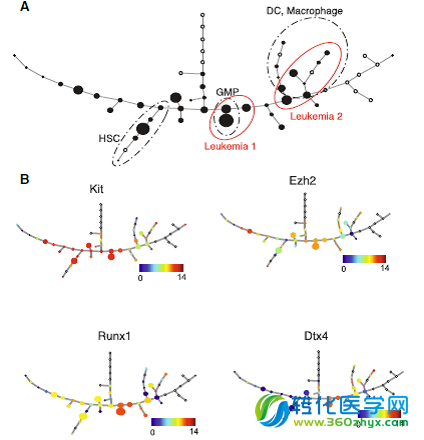
研究团队利用Fluidigm的微流控系统,对71个白血病单细胞和190个正常骨髓细胞的175个基因的mRNA水平进行了检测。为了找到白血病细胞间异质性,研究人员运用SPADE降维算法对单细胞的定量PCR结果进行分析,并对分析结果用二位的树形图进行展示。结果显示白血病细胞能分为两个类群,两群细胞在SPADE树的不同分支上,第一群包含29个单细胞,转录组更接近于单核粒细胞前体,第二群包含42个单细胞,转录组更接近于树突状细胞和巨噬细胞(图2)。他们用Mann-Whitney检测方法比较了第一类群白血病细胞和单核粒细胞前体的基因表达水平,发现Meis1,Cdkn2c等4个基因的在第一类白血病细胞中的表达水平显著高于单核粒细胞前体,说明这一亚群的细胞具有更强的侵染增殖能力。随后,他们比较了两个白血病亚群细胞之间的基因表达水平,发现第一类亚群细胞中Kit,Etv6,Runx1,Suz12,Ezh2,Brd3等基因的表达水平高于第二类。
最后通过集落形成实验(colony-forming assays),研究团队发现第一类白血病细胞的增殖能力显著高于第二类白血病细胞。白血病肿瘤干细胞更可能蕴藏在第一类白血病细胞中。
“单细胞基因表达谱数据和高通量数据分析手段的结合,我们可以从全新的视角剖析肿瘤细胞的异质性,找到肿瘤干细胞的标志基因,加速新抗瘤治疗手段的诞生,使治疗预后更理想。”作者写道。
英国剑桥大学欧洲生物信息中心的Oliver团队,创建了一种名为scLVM的数学模型,并用该方法对正在分化为Th2细胞的T细胞的单细胞RNA-seq数据进行分析,发现了分化过程中的细胞类群差异,首次揭示了T细胞向Th2细胞分化过程中的两个阶段(ref. 3)。
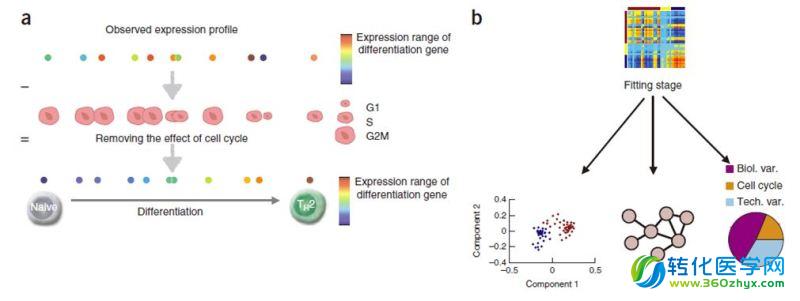
Th2细胞是第二类辅助型T细胞,在调控感染,过敏,肿瘤免疫,妊娠反应等方面都有非常重要的作用。研究人员在天然T细胞向Th2细胞分化的过程中,利用Fluidigm C1系统捕获制备了96个单细胞的cDNA,并进行了单细胞RNA-Seq实验。对数据进行质控分析后得到81个单细胞7073个差异表达的基因数据。研究人员发现,2881个(44%)基因的表达水平会受到细胞周期的影响。这些伴随细胞周期阶段变化而产生的基因表达水平的改变,就像一个较高的“背景”,会掩盖分化过程中与单细胞异质性相关的基因表达差异。
“因此我们建立了一个新的数学模型,scLVM,它可以在数据分析时,剔除细胞周期给基因表达带来的影响”,作者说道。“这个数学模型会根据单细胞所处的有丝分裂时期,如G1、S或G2/M,对其基因的表达值进行校正。我们可以在剔除细胞周期干扰的条件下,研究单细胞间基因表达的差异和细胞异质性。“(图4)
通过scLVM数学模型的分析,研究人员发现,7073个差异表达基因中,至少有27%的基因,其单细胞间的表达差异主要是由细胞所处分裂周期阶段的不同导致的。在剔除这些细胞周期因素的干扰后,研究团队根据校正的基因表达数据进行组成分分析(PCA),他们惊奇的发现,在向Th2细胞分化的过程中,T细胞可以清晰的分为两大类,其中一类 Il4ra35,Gata3,Stat3, Klf13,Batf,Il24等Th2细胞标志基因的表达水平显著高于另一类,说明这一类代表了完全成熟的Th2细胞,而另一类则是非成熟的Th2细胞。结果也说明了T细胞向Th2细胞分化的过程要经历两个阶段。研究人员分析校正后的基因表达数据,发现了这两类细胞存在401个差异表达的基因,若用校正前的数据只能找到7个基因。
这个案例通过建立scLVM的算法模型,剔除了细胞周期等因素的干扰,使科研工作者更容易发现隐藏的细胞异质性和细胞类群。也说明在单细胞异质性的研究过程中,还面临着很多新的挑战,需要创建新的研究方法才能得以完满解决。
Reference:
1.Ennen, M. et al. “Single-cell gene expression signatures reveal melanoma cell heterogeneity.” Oncogene 34(25) (2015): 3,251-63.
2.Saadatpour, A. et al. “Characterizing heterogeneity in leukemic cells using single-cell gene expression analysis.” Genome Biology 15(12) (2014): 525.
研究细胞异质性,是一个单细胞层面的范畴。单细胞间的异质性存在于DNA、RNA、蛋白等各个层面。在此,我们将结合具体案例,介绍从单细胞转录组着手,通过高通量数据分析方法揭示单细胞异质性的研究思路。
单细胞基因表达谱,叩开细胞异质性大门目前,通过高通量的分子技术平台,对一定数量的单细胞的进行基因表达水平的检测(RNA或蛋白),是揭示细胞异质性最有效的途径之一。
法国国家科研中心分子细胞及遗传学研究所I Davidson团队通过对黑色素瘤单细胞进行基因表达水平的研究,发现了黑色素瘤两个亚类细胞群(增殖型细胞和侵染(invasive)型细胞)之间异质性产生的分子基础,并为临床肿瘤检测提供了全新的思路和线索(ref. 1)。
肿瘤并不是均一的细胞团,肿瘤组织的不同细胞具有不同的侵染增殖能力及成瘤潜能。传统的抗肿瘤药物对增殖速度快的肿瘤细胞清除效果较好,但对已发生转变并获得耐药性的细胞则效果十分有限。I Davidson研究团队将一株增殖速度快但侵染能力差的黑色素瘤细胞注入小鼠皮下培养成瘤,待12周后将实体瘤取出,利用Fluidigm C1全自动单细胞制备系统获得该肿瘤单细胞的cDNA样本,并利用Biomark高通量qPCR仪对92个单细胞的113个基因进行了检测。引人注目的是,他们在少量肿瘤单细胞中检测出了侵染(invasive)相关基因(ZEB1,GLI2,MYOF)和耐药基因(ABCBS)的表达(图1)。
“我们利用单细胞的检测手段,发现在原本低侵染性的黑色素瘤细胞中,一小部分已经转变成了高侵染性的肿瘤细胞,并且还具备了耐药性。”作者说道。“相比起传统的活检手段,针对肿瘤单细胞进行分析,能更好的揭示肿瘤异质性,挖掘新的肿瘤标记,揭示肿瘤形成的过程。”
图1. 92个肿瘤单细胞中113个基因表达水平的聚类热图
大数据研究思维,挖掘隐藏的单细胞差异
除了高通量的实验技术,研究单细胞异质性还要综合运用各种大数据的分析方法。美国Dana-Farber肿瘤研究中心的Guo-Cheng Yuan团队综合运用多种高通量数据的分析手段,首次发现白血病细胞可以分为两个亚群,而且白血病肿瘤干细胞可能匿藏于其中一个亚群中(ref. 2)。
研究团队利用Fluidigm的微流控系统,对71个白血病单细胞和190个正常骨髓细胞的175个基因的mRNA水平进行了检测。为了找到白血病细胞间异质性,研究人员运用SPADE降维算法对单细胞的定量PCR结果进行分析,并对分析结果用二位的树形图进行展示。结果显示白血病细胞能分为两个类群,两群细胞在SPADE树的不同分支上,第一群包含29个单细胞,转录组更接近于单核粒细胞前体,第二群包含42个单细胞,转录组更接近于树突状细胞和巨噬细胞(图2)。他们用Mann-Whitney检测方法比较了第一类群白血病细胞和单核粒细胞前体的基因表达水平,发现Meis1,Cdkn2c等4个基因的在第一类白血病细胞中的表达水平显著高于单核粒细胞前体,说明这一亚群的细胞具有更强的侵染增殖能力。随后,他们比较了两个白血病亚群细胞之间的基因表达水平,发现第一类亚群细胞中Kit,Etv6,Runx1,Suz12,Ezh2,Brd3等基因的表达水平高于第二类。
图2. 白血病细胞的SPADE分型展示图
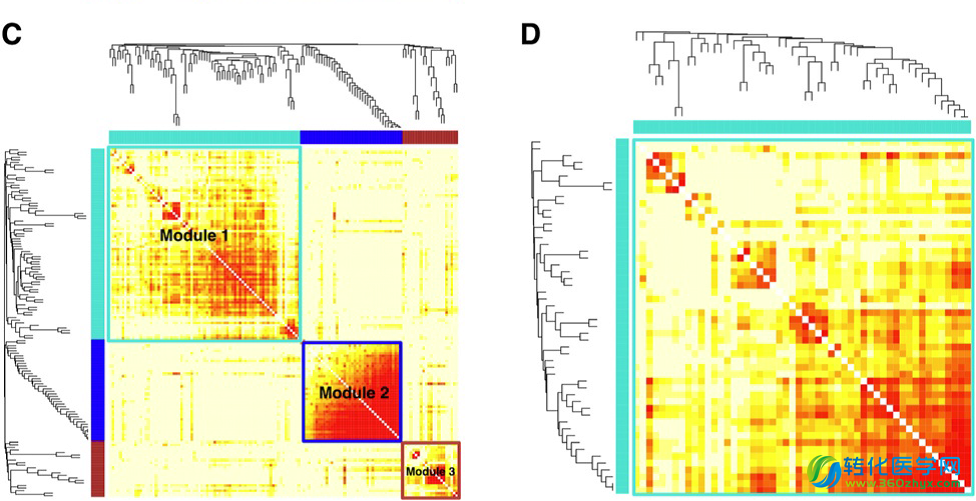
除了基因的表达差异,单细胞间的异质性也体现在基因调控网络的差异上。WGCNA的分析方法,可将具有表达相关性的基因聚类成模块(module),揭示在特定细胞群中基因间的表达调控关系。研究人员利用该方法发现,第一类白血病细胞和第二类白血病细胞的基因共表达网络(co-expression network)模块差别很大。一类白血病细胞有三大共表达基因模块,而二类白血病细胞只有一个模块。一类白血病细胞模块2中的40个基因,只有5个与二类白血病模块中的基因相同,说明两类白血病细胞的在基因调控关系上也存在显著差异(图3)该结论还用另一种称为DiffCoEx的研究方法进行了验证。最后通过集落形成实验(colony-forming assays),研究团队发现第一类白血病细胞的增殖能力显著高于第二类白血病细胞。白血病肿瘤干细胞更可能蕴藏在第一类白血病细胞中。
“单细胞基因表达谱数据和高通量数据分析手段的结合,我们可以从全新的视角剖析肿瘤细胞的异质性,找到肿瘤干细胞的标志基因,加速新抗瘤治疗手段的诞生,使治疗预后更理想。”作者写道。
图3. WGCNA方法解析两类白血病细胞的共表达网络模块
新颖的分析手段,突破研究的瓶颈
在具体研究过程中,有一些干扰因数,可能会阻碍我们发现隐藏在单细胞间的异质性,比如细胞周期对基因表达的影响,就会严重干扰常规统计算法对细胞异质性的评估。因此量体裁衣,根据具体情况创建新的分析方法也尤为重要。英国剑桥大学欧洲生物信息中心的Oliver团队,创建了一种名为scLVM的数学模型,并用该方法对正在分化为Th2细胞的T细胞的单细胞RNA-seq数据进行分析,发现了分化过程中的细胞类群差异,首次揭示了T细胞向Th2细胞分化过程中的两个阶段(ref. 3)。
Th2细胞是第二类辅助型T细胞,在调控感染,过敏,肿瘤免疫,妊娠反应等方面都有非常重要的作用。研究人员在天然T细胞向Th2细胞分化的过程中,利用Fluidigm C1系统捕获制备了96个单细胞的cDNA,并进行了单细胞RNA-Seq实验。对数据进行质控分析后得到81个单细胞7073个差异表达的基因数据。研究人员发现,2881个(44%)基因的表达水平会受到细胞周期的影响。这些伴随细胞周期阶段变化而产生的基因表达水平的改变,就像一个较高的“背景”,会掩盖分化过程中与单细胞异质性相关的基因表达差异。
“因此我们建立了一个新的数学模型,scLVM,它可以在数据分析时,剔除细胞周期给基因表达带来的影响”,作者说道。“这个数学模型会根据单细胞所处的有丝分裂时期,如G1、S或G2/M,对其基因的表达值进行校正。我们可以在剔除细胞周期干扰的条件下,研究单细胞间基因表达的差异和细胞异质性。“(图4)
通过scLVM数学模型的分析,研究人员发现,7073个差异表达基因中,至少有27%的基因,其单细胞间的表达差异主要是由细胞所处分裂周期阶段的不同导致的。在剔除这些细胞周期因素的干扰后,研究团队根据校正的基因表达数据进行组成分分析(PCA),他们惊奇的发现,在向Th2细胞分化的过程中,T细胞可以清晰的分为两大类,其中一类 Il4ra35,Gata3,Stat3, Klf13,Batf,Il24等Th2细胞标志基因的表达水平显著高于另一类,说明这一类代表了完全成熟的Th2细胞,而另一类则是非成熟的Th2细胞。结果也说明了T细胞向Th2细胞分化的过程要经历两个阶段。研究人员分析校正后的基因表达数据,发现了这两类细胞存在401个差异表达的基因,若用校正前的数据只能找到7个基因。
这个案例通过建立scLVM的算法模型,剔除了细胞周期等因素的干扰,使科研工作者更容易发现隐藏的细胞异质性和细胞类群。也说明在单细胞异质性的研究过程中,还面临着很多新的挑战,需要创建新的研究方法才能得以完满解决。
图4. scLVM数学模型概述
Reference:
1.Ennen, M. et al. “Single-cell gene expression signatures reveal melanoma cell heterogeneity.” Oncogene 34(25) (2015): 3,251-63.
2.Saadatpour, A. et al. “Characterizing heterogeneity in leukemic cells using single-cell gene expression analysis.” Genome Biology 15(12) (2014): 525.
3.Buettner, F. et al. “Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells.” Nature Biotechnology 33(2) (2015): 155-60.
(转化医学网360zhyx.com)
还没有人评论,赶快抢个沙发