 腾讯登录
腾讯登录基因诊疗时代,排查儿童的隐形杀手!
| 导读 | 点击上方“转化医学网”订阅我们!干货|靠谱|实用 池塘边的榕树上知了在声声叫着夏天操场边的秋千上只有蝴蝶停在上面黑板上老师地粉笔还在拼命唧唧喳喳写个不停等待着下课等待着放学等待游戏的童年每当听到罗大佑的这首童年就会勾起无限遐想。小伙伴的嬉笑打闹声,刚修剪过的草坪青草香味,放学后外婆递来的雪糕香甜味,无忧无虑的孩童时代让我很庆幸能有一个快乐的童年,要知道对于一些得了罕见病的儿童,这 |
池塘边的榕树上
知了在声声叫着夏天
操场边的秋千上
只有蝴蝶停在上面
黑板上老师地粉笔
还在拼命唧唧喳喳写个不停
等待着下课
等待着放学
等待游戏的童年
每当听到罗大佑的这首童年就会勾起无限遐想。小伙伴的嬉笑打闹声,刚修剪过的草坪青草香味,放学后外婆递来的雪糕香甜味,无忧无虑的孩童时代让我很庆幸能有一个快乐的童年,要知道对于一些得了罕见病的儿童,这样的童年是不可想象的。
精准医学拯救地贫儿童
出生在美国伊利诺伊州的小女孩旺达·西哈纳(Wanda Sihanath)是一个爱笑充满活力的女孩,小时候她被父母送去学芭蕾舞。在芭蕾舞的训练中旺达·西哈纳发现自己很容易疲劳,很快她在训练中就病倒了。经过诊断,医生确诊她得了β-地中海贫血症。
地中海贫血症这种疾病的标准治疗方法是使用基因型相近的亲属的骨髓移植——通常需要亲兄弟姐妹。但是,大多数人都无法找到可以做骨髓移植的供体。即使能找到基因型相近供体,发生免疫排斥等严重副反应的几率也很高。这意味着西哈纳每个月都需要到医院输血才能维持正常生活。
直到2014年,旺达·西哈纳参加了一项临床试验,成为了第一位受试者。在这项试验中,医生把她的骨髓干细胞从体内分离出来,并且用一种经过改造的病毒把正常的HBB基因“输入”到这些细胞。然后,她再一次接受了输血。不同的是,这次输入的是含有她自己骨髓干细胞的血液。
神奇的事情发生了。自这次输血后,西哈纳再也没有输过血。她的身体恢复了正常,就像没有得过这种遗传病一样。
地中海贫血症
β-地中海贫血症是一种罕见的遗传性血液疾病,由一个叫做HBB的基因发生突变而产生。这个基因负责产生血红蛋白,而血红蛋白是我们血液中运输氧气的红细胞的主要成分。当HBB基因发生了突变而不能产生正常的血红蛋白,就会导致溶血和严重贫血。
全球每年约有790万出生缺陷患儿,其中5种常见疾病占7000种出生缺陷的25%,地中海贫血的发病率位于第三位。地中海贫血可分为α地中海贫血和β地中海贫血,重型α地贫患儿严重水肿不能存活,重型β于贫患儿出生后需要通过去铁、脾脏切除、长期输血等综合治疗,重型β地中海贫血患儿目前还没有根治的办法,因此寿命及生存质量不高。在我国地中海贫血以南方地区多见,尤以广西、广东和海南最多。
地中海贫血是常染色体隐性遗传病,那么地中海贫血的遗传概率有多大?轻型地贫携带者同正常人婚配,其后代有 50%的机会成为轻型地贫携带者。静止型地贫与轻型地贫婚配,有 1 /4机会生出地中海贫血患儿。如果如果夫妻为同型地贫基因携带者,每次怀孕,胎儿有1/4的机会为正常,1/2的机会为基因携带者,另1/4的机会为重型地中海型贫血患者,而如果夫妻双方携带的是不同型的地贫基因,或者只有一方携带地贫基因,所生的孩子不会得地贫。
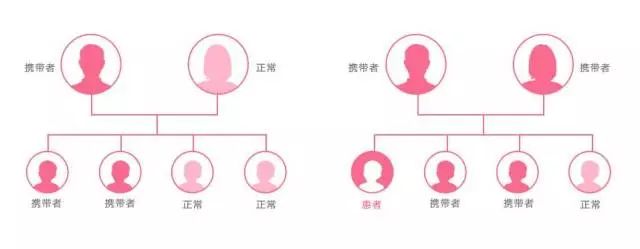
罕见疾病筛查梳理
对于罕见疾病尚无有效的治疗方法,超过95%的罕见病没有FDA或EMA通过的上市药物,约80%的罕见病是由基因引起的。只能广泛开展产前筛查与产前诊断,及时诊断胎儿异常,适时终止妊娠,降低出生缺陷。关于产前筛查可以看之前写的关于NIPT的文章母乳喂养日,我想对你说520。
唐氏(Down)综合征(T21染色体异常):唐氏(Down)综合征又称21三体综合征,是一种最常见的染色体数目异常,在活产新生儿中发病率约为1/700~1/800,患者核型可分为单纯三体型、易位型及嵌合型,绝大多数患者为单纯三体型。60%患儿在胎内早期即流产,存活者有明显的智能落后、特殊面容、生长发育障碍和多发畸形。
爱德华氏(Edward)综合征(T18染色体异常):爱德华氏(Edward)综合征又称18三体综合征,是发生率仅次于唐氏综合征的染色体异常疾病,临床表现为严重的神经系统发育障碍、面部畸形、生长迟缓、骨骼异常、心脏和肾脏畸形等,存在严重的智力低下和多种缺陷,新生儿发生率约为1/5000。
帕陶氏(Patau)综合征(T13染色体异常):帕陶氏(Patau)综合征又称13三体综合征,是常见染色体异常疾病之一,常引起胎儿严重的多发结构畸形,包括颅脑、颜面部及心脏畸形等,同时存在严重的智力发育障碍,新生儿中发病率约为1/25000,女性明显多于男性。
特纳(Turner)综合征(45,X):特纳(Turner)综合征又称先天性卵巢发育不全,是一种先天性染色体异常所致的疾病,由于父体或母体在减数分裂过程中发生变异,使一方带X染色体的生殖细胞与另一方不带性染色体或畸变的X染色体的生殖细胞结合而发病,临床特点为身矮、生殖器和第二性征不发育及躯体发育异常,发生率为新生儿的10.7/10万或女婴的22.2/10万。
克兰费尔特(Klinefelter)综合征(47,XXY):克兰费尔特(Klinefelter)综合征又称先天性曲细景精管发育不全综合征,是一种较为常见的性染色体畸变遗传病,是由于父母的生殖细胞在减数分裂形成精子和卵子的过程中,性染色体发生不分离现象所致,患者性染色体为47,XXY,即比正常男性多了1条X染色体,临床表现为患者有类无睾身材、男性乳房发育、小睾丸、无精子及尿中促性腺激素增高等。
单基因遗传疾病:单基因遗传病是指受一对等位基因控制的遗传病,有6600多种,并且每年在以10-50种的速度递增,单基因遗传病已经对人类健康构成了较大的威胁,在我国,地中海贫血、杜氏肌营养不良症、甲型血友病和先天性软骨发育不全是常见的单基因遗传病。
地中海贫血:地中海贫血(简称地贫)是我国南方各省最常见、危害最大的遗传病,人群发生率高达10%以上,以广东、广西为主。地中海贫血分为α型、β型、δβ型和δ型4种,其中以β和α地中海贫血较为常见。本病的发生是由于血红蛋白分子中的珠蛋白肽链结构异常或合成速率异常,造成肽链不平衡而产生以溶血性贫血为主的症状群。通过不同的基因诊断方法已能对16种常见β地贫基因和6种常见α地贫基因进行诊断,即能对占我国人群90%以上的地贫基因作出诊断。
杜氏肌营养不良症:杜氏肌营养不良症是由于肌营养不良蛋白基因缺陷而导致的一种X染色体隐形遗传疾病,通常男性发病,女性多为杂合子携带者,发病率约为活产男婴的1/3500。该病是一种致死性神经肌肉性疾病,患者常于3~5岁发病,预后差,目前尚无有效治疗方法,因此进行产前诊断和遗传咨询是防止患儿出生最重要的措施。
甲型血友病:甲型血友病是由于凝血因子VIII的遗传缺陷或缺乏导致凝血活酶生成障碍的一种常见X染色体隐形遗传疾病,约占先天性出血性疾病的85%,该病主要是女性传递,男性发病,多见隔代遗传,男性患病率约为1/5000,约60%患者有遗传病家族史。
先天性软骨发育不全:先天性软骨发育不全是一种由于软骨内骨化缺陷的先天性发育异常,是一种以短肢、躯干相对正常和巨头为特征的常染色体显性遗传性侏儒,常合并有其它遗传性疾病或出现肌肉骨骼系统其他畸形以及呼吸、神经系统的严重并发症,在我国的发生率为18/1000000。
以上列举的基因遗传疾病都可以通过基因检测技术筛查,达到疾病预防的结果,有助于控制新生儿罕见病发病,减少出生缺陷。随着科学的进展,基因疗法也在全球范围内取得了一系列突破性进展,人们也认识到基础研究对基因治疗的重要性超过以往任何治疗手段。

有收获 记得 转发、分享 让更多的朋友知道哦
(转化医学网360zhyx.com)

-
游客2018-06-29 13:54:55好
 正在加载
正在加载