 腾讯登录
腾讯登录【独家】2018上半年FDA批准的17个新药汇总及逐个评述
| 导读 | 2018年上半年,FDA药品审评与研究中心(CDER)批准了17个新药,这些新药包括新药申请NDA中的新分子实体(NME)和生物制品许可申请(BLA)。 |
2018年上半年,FDA药品审评与研究中心(CDER)批准了17个新药,这些新药包括新药申请NDA中的新分子实体(NME)和生物制品许可申请(BLA)。
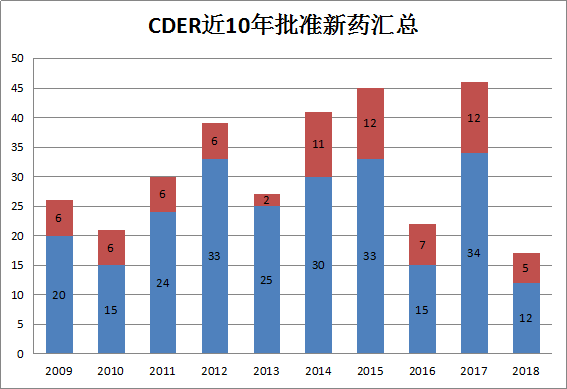
上图是过去10年中,CDER每年批准的新药数。2018年上半年CDER批准的新药包括5个BLA和12个NME。相比于2017年上半年的23个减少了6个,略高于2016上半年和2015上半年。
罕见病/孤儿药
(Rare OR "Orphan" Diseases)
已批准的17个新药中,8个新药获得了孤儿药资格(O),占CDER批准新药的47%。罕见病有两个特征,其一为患者人数少,美国为少于20万人的疾病,欧盟属于5/10000的疾病;其二为危及生命和健康的严重疾病。
优先审评(Priority Review)
如果CDER确定药品能够有潜力对医疗保健做出实质性推动,药品将获得优先审评。药品在6个月内而不是标准的10个月内审评。2018年上半年获批新药中有8个被认定为优先审评(P),占17个新药的47%。
CDER应用多种监管方法加快新药研发和审批。这些方法除了优先审评还包括:快速通道(Fast Track)、突破性治疗认定(Breakthrough)和加速批准(Accelerated Approval)。
治疗领域方面,2018年上半年还是比较丰富,抗肿瘤药明显减少,仅有2个。整体来说,美国FDA还是各药企新药上市的首选,17个新药有12个药物都是全球首批,其余的大多在欧盟首批。
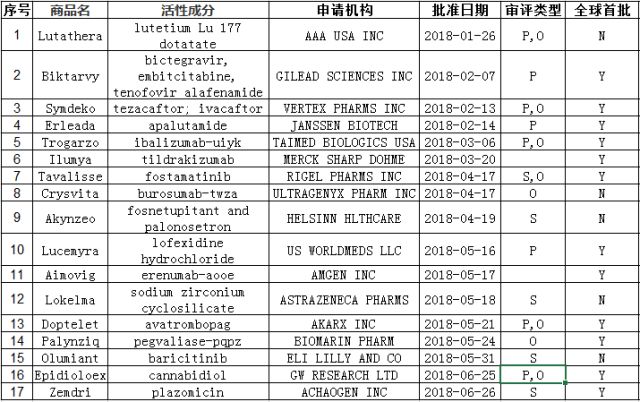
下表为2018年批准的17个新药汇总,来源于FDA网站,Novel Drug Approvals for2018。
品种评述
01 Lutathera
(lutetium Lu 177 dotatate)
1月26日,FDA批准了诺华子公司法国Advanced Accelerator Applications 公司Lutathera用于治疗影响胰腺或胃肠道的一类癌症,即胃肠胰腺神经内分泌肿瘤(GEP-NETs)。
Lutathera是一种Lu-177标记的生长抑素类似物,属于新兴的肽受体放射性核素疗法(PRRT),通过与一种称为生长激素抑制素受体的细胞结合而起作用,该生长抑素受体可能存在于某些肿瘤上。在与受体结合之后,药物进入细胞,释放辐射来损伤肿瘤细胞。
在美国和欧盟,Lutathera均被授予孤儿药地位。欧盟已于2017年10月批准用于治疗不可切除或转移的生长抑素受体阳性胃肠胰腺神经内分泌肿瘤成人患者。这也是放射性药物首次被FDA批准用于治疗GEP-NETs。
02 Biktarvy
(bictegravir, embitcitabine,
tenofovir alafenamide)
2月7日,FDA批准了Gilead公司的Biktarvy,作为每日一次的单片片剂疗法,用于治疗HIV-1感染。
Biktarvy是一款全新的无助推(unboosted)整合酶链转移抑制剂(INSTI),由bictegravir(50mg)、emtricitabine(200mg)、与tenofovir alafenamide(25mg)三种成分组成。
Biktarvy的疗效与安全性在4项正在进行的3期临床试验中得到了验证。试验1489和1490招募的是初治的HIV-1感染成人患者,而试验1844与1878则招募了病毒感染在病毒学上得到抑制的成人患者。
在试验1489中,629名患者以1:1的比例分为两组,分别接受Biktarvy与abacavir/dolutegravir/lamivudine(600/50/300mg)的治疗。在48周后,这两组患者中,分别有92.4%和93.0%的患者达到了HIV-1 RNA小于每毫升50c的主要终点。而在试验1490中,645名患者接受的分别是Biktarvy与dolutegravir/FTC/TAF。同样,两组达到主要终点的比例接近,分别为89.4%与92.9%。
在试验1878中,577名在药物作用下,HIV-1 RNA已经小于每毫升50c的成人患者被1:1分为两组,一组继续现有的疗法,另一组则切换到Biktarvy的治疗。在48周后,两组中均有1.7%的患者其HIV-1 RNA回到不低于每毫升50c的水平;而根据FDA的算法,分别有92.1%(Biktarvy组)与88.9%(现有疗法组)的患者保持了HIV-1 RNA小于每毫升50c。
03 Symdeko
(tezacaftor; ivacafto)
2月13日,FDA批准了Vertex医药公司的Symdeko上市,用于治疗12岁及以上的囊性纤维化(cystic fibrosis,CF)患者。SYMDEKO是Vertex获得FDA批准的第3种针对囊性纤维化根本病因的治疗药物。
囊性纤维化是一种罕见的会缩短寿命的遗传疾病,影响了北美、欧洲和澳大利亚约 75000 名患者,由基因突变导致的 CFTR 蛋白缺陷或缺失引起。儿童必须遗传到两个有缺陷的 CFTR 基因——分别来自父母,才会患囊性纤维化。CFTR 基因中有约 2000 个已知的突变,其中一些突变会通过在细胞表面产生不工作或过少的 CFTR 蛋白,它们可以通过基因检测或基因分型检测来确定。CFTR 蛋白的功能缺陷或缺失导致许多器官细胞中盐和水的流入和流出不均衡。在肺中,这会造成异常粘稠的粘液积聚,引起慢性肺部感染和进行性肺损伤,并最终导致死亡。囊性纤维化患者的中位寿命在 20 岁左右。
据悉,欧洲药品管理局(EMA)已经确认了tezacaftor/ivacaftor组合药物的营销授权申请(MAA)。公司预计在2018年下半年获得欧盟批准。
04 Erleada(apalutamide)
2月14日,FDA批准强生Erleada (apalutamide) 上市,用于治疗非转移性(前列腺癌细胞未扩散)去势抵抗(激素治疗后疾病仍进展)的前列腺癌。
Erleada获得过FDA的优先审评资格,是FDA批准的首个治疗非转移性去势抵抗前列腺癌的药物,也是首个凭借无转移生存期(metastasis-free survival,MFS)的临床终点获批上市的肿瘤新药。
Erleada的安全性及疗效在涉及1207例非转移性去势抵抗前列腺癌的随机研究中得到证实。患者随机给予apalutamide或安慰剂,并接受内分泌治疗,包括接受促性腺激素释放激素(GnRH)类似物,或者手术去势(切除双侧睾丸)以降低体内雄激素水平。结果显示,apalutamide治疗组无转移生存期相比安慰剂组显著延长(40.5 vs 16.2个月)。
05 Trogarzo(ibalizumab-uiyk)
2018年3月7日,美国FDA宣布正式批准药明生物合作伙伴中裕新药(TaiMed)的Trogarzo(ibalizumab-uiyk)上市,作为一种全新的抗逆转录病毒疗法,治疗现有多种疗法均无法起效的成人HIV感染者。值得一提的是,这是美国FDA在2018年批准的首款创新生物药,此项目也是药明生物首个商业化生产的项目,标志着药明生物跻身成为全球少数几个通过FDA GMP认证的生物药合作研发生产服务商(CDMO)。
Trogarzo是一种静脉滴注的人源化免疫球蛋白G4单克隆抗体,它与CD4+ T细胞受体的第二个胞外区域结合,阻止HIV病毒入侵这些细胞。
Trogarzo的安全性与疗效在一项临床试验中得到了验证。该试验招募了40名感染有多重耐药性HIV的患者,他们均重度经治,有些患者甚至已经接受过10种或更多的抗逆转录病毒疗法。然而即便接受了大量治疗,他们血液中的病毒水平(HIV-RNA)依旧很高。研究人员发现,在现有的疗法中额外加入Trogarzo的治疗后,只要短短一周,大部分患者血液中的HIV-RNA水平就有显著下降。24周后,43%的患者其HIV-RNA水平依旧得到了抑制。对于这些急缺治疗方案的患者,Trogarzo带来了显著益处。
06 Ilumya(ibalizumab-uiyk)
3月20日,FDA批准ILUMYA (tildrakizumab-asmn)上市,用于适合接受全身治疗或光疗的中重度斑块型银屑病成人患者。
据3期临床试验数据显示,与安慰剂相比,施用ILUMYA 100 mg能取得显著的临床改善。研究通过两次用药后第12周至少75%的皮肤清除率(银屑病面积敏感指数或PASI 75),以及医师全面评估(PGA)评分“清除”或“最小”进行测量。其中,该研究有74%(229人)的患者在三次用药后第28周达到75%的皮肤清除率,84%持续用药的患者在第64周能维持PASI 75。毋庸置疑,ILUMYA是中度至重度斑块型银屑病临床治疗上的又一个重要突破。
07 Tavalisse(fostamatinib)
4月17日,美国FDA批准了Rigel制药的TAVALISSE用于对之前治疗缓解不佳的成年慢性免疫性血小板减少症(ITP)患者血小板减少的治疗。TAVALISSE是一种口服的脾脏酪氨酸激酶(SYK)抑制剂,通过阻止血小板的破坏来应对疾病的潜在自身免疫原因,为成年慢性ITP患者提供了一个重要的新的治疗方案。
TAVALISSE的批准依赖于FIT临床研究项目的数据,FIT包含两项随机安慰剂对照的临床3期研究047和048、一项开标扩展试验049以及最初的概念验证试验。新药上市申请包括了163例ITP患者的数据,并得到了一项安全数据集的支持,该数据集包含4600多名涉及其它适应症的已接受TAVALISSE评估的受试者。
08 Crysvita(burosumab-twza)
4月17日,美国FDA批准了Ultragenyx Pharmaceutical公司的新药Crysvita,成为首个获批治疗1岁及以上儿童和成年人的X连锁低磷血症(XLH)的药物。XLH是一种罕见的遗传性软骨病,会造成血液中磷含量低,导致儿童和青少年的骨骼生长和发育受损,并使其一生都有骨矿化的问题。
Crysvita获批是基于4项临床试验中的安全性和疗效数据。在一项安慰剂对照临床试验中,94%的每月接受一次Crysvita的成年人能达到正常的磷水平,而安慰剂组患者只有8%能达到这一水平。在儿童中,每两周接受一次Crysvita的患者中有94%到100%能达到正常的磷水平。在儿童和成人中,接受Crysvita疗法的患者的X光(与XLH相关)结果有所改善。把这些结果与自然历史队列相比较,也为Crysvita的有效性提供了支持。
09 Akynzeo (fosnetupitant
and palonosetron)
4月19日,FDA批准了瑞士制药集团Helsinn公司的静脉注射AKYNZEO,作为经历CINV的患者的替代治疗选择。
FDA已批准将AKYNZEO®IV与成人地塞米松联合用于预防与高度致吐性癌症化疗的初始和重复过程有关的急性和延迟性恶心和呕吐。尚未研究AKYNZEO®用于预防与蒽环类加环磷酰胺化疗相关的恶心和呕吐
口服AKYNZEO在2014年被美国食品药物管理局批准为固定组合口服药,用于预防与癌症化疗初期和重复过程有关的急性和延迟性恶心和呕吐,包括但不限于高度致吐化疗。
AKYNZEO®静脉注射制剂的批准为将这种重要治疗选择带入更多患者采用新配方铺平了道路,并将于2018年5月在美国推出此产品。
10 Lucemyra
(ofexidine hydrochloride)
5月16日,美国FDA宣布批准US WorldMeds的Lucemyra(盐酸洛非斯汀,lofexidine hydrochloride),用于缓解突然停用阿片类药物的成人患者的戒断症状。虽然Lucemyra可以减轻戒断症状的严重程度,但它无法完全阻止这些症状,并且最长只能使用14天。
Lucemyra的安全性和有效性得到了两项随机、双盲、安慰剂对照临床试验的支持。这些临床试验共有866名符合阿片类药物依赖诊断标准的成人患者,这些患者在生理上依赖于阿片类药物,并正在进行阿片类药物戒断。这些研究使用Gossop短暂停药量表(SOWS-Gossop)来评估疗效,这是一种通过患者报告结果来评估阿片类戒断症状的工具。这些症状包括感觉不适、胃痉挛、抽搐、感觉冷、心脏剧烈跳动、肌肉紧张、疼痛,打呵欠、流泪和失眠。
11 Aimovig(erenumab-aooe)
5月17日,FDA批准了安进(Amgen)公司的Aimovig,作为成人偏头痛的预防性治疗,给药方式为每月一次的自我注射,这也是FDA批准的首个预防性偏头痛治疗药物。
Aimovig获批主要基于围绕其展开的三项临床试验,试验评估了其在偏头痛预防中的安全性和有效性。
第一项研究包括955名有发作性偏头痛病史的参与者,并将Aimovig与安慰剂进行比较。在6个月的时间里,aimovig治疗的患者平均每月的偏头痛天数比服用安慰剂的患者少1到2个月。
第二项研究包括577名有发作性偏头痛病史的患者,并将Aimovig与安慰剂进行比较。在三个月的时间里,aimovig治疗的患者平均每个月比服用安慰剂的病人少一次偏头痛。
第三项研究对667例慢性偏头痛患者进行了评估,并将Aimovig与安慰剂进行比较。在这项研究中,在三个月的时间里,接受Aimovig治疗的患者平均每月比接受安慰剂的患者少2次。临床试验中最常见的副作用是注射部位的反应和便秘。
12 Lokelma (sodium zirconium cyclosilicate)
5月18日,美国FDA批准了阿斯利康(AstraZeneca)的新药Lokelma用于治疗罹患高钾血症(hyperkalaemia)的成人患者。
高血钾风险在慢性肾病(CKD)患者以及服用普通心力衰竭(HF)药物(如肾素-血管紧张素-醛固酮系统[RAAS]抑制剂)的患者中显著增加,因为这种药物会增加血钾水平。
Lokelma于今年3月底在欧盟获批上市。次Lokelma获得FDA的批准是基于三项双盲、安慰剂对照试验和两项开放标签试验数据的支持。
这些研究表明,Lokelma的起效时间是服药后1.0小时,达到正常血钾水平的中位时间是2.2小时,92%的患者在基线后48小时内达到正常血钾水平。该药物的治疗效果可维持长达12个月。
13 Doptelet(avatrombopag)
5月21日,美国FDA批准Dova Pharmaceuticals子公司AkaRx的新药Doptelet片剂,用于治疗计划接受医疗或牙科手术的慢性肝病(CLD)成人患者的低血小板计数(血小板减少症)。
Doptelet的安全性和有效性在两项试验(ADAPT-1和ADAPT-2)中得到了验证。这些研究共包含435名慢性肝病和严重血小板减少症患者,他们将接受通常需要输注血小板的手术。这些试验评估了两个剂量水平的口服Doptelet与安慰剂相比治疗5天的效果。结果显示,与安慰剂组相比,两种剂量水平的Doptelet组有较高比例的患者具有增加的血小板计数,并且不需要在手术当天和治疗后7天内接受血小板输注或任何救援治疗。Doptelet最常见的副作用有发烧、胃(腹)痛、恶心、头痛、疲劳和手足肿胀(水肿)。
14 Doptelet(avatrombopag)
5月24日,FDA批准了BioMarin Pharmaceutical公司的Palynziq注射剂,用于降低苯丙酮尿症(PKU)成人患者的血液苯丙氨酸(Phe)水平 ,这些患者在现有管理下其血液Phe浓度无法控制在600微摩尔/升以内。
Palynziq是一种聚乙二醇化的重组苯丙氨酸解氨酶,用来替代PKU患者缺乏的苯丙氨酸羟化酶(PAH)以分解Phe。在关键3期研究PRISM-2中,Palynziq与安慰剂相比显著降低了血液Phe水平(p<0.0001),抵达了血液Phe变化的主要终点。在PRISM-2双盲、安慰剂对照、随机停药期试验(RWP)中,患者以2:1的比例被随机分配继续接受Palynziq治疗(每日20 mg或每日40 mg)或接受安慰剂,持续8周。结果显示,Palynziq组患者可以维持血液Phe浓度,而安慰剂组患者的血液Phe浓度恢复到治疗前基线。
15 Olumiant(baricitinib)
5月31日,美国FDA批准了礼来公司(Eli Lilly)新药Olumiant上市,治疗罹患中度至重度类风湿关节炎,却无法从TNF抑制剂治疗中受益的成人患者。
由礼来与Incyte带来的Olumiant就是这样一款充满潜力的新药。它是一款每日一次的口服JAK抑制剂,能高效抑制JAK1、JAK2、以及TYK2。在人体内,不少细胞因子依赖于JAK的活性,其在不少炎性疾病和自身免疫疾病的发病过程中有潜在作用。通过抑制多种JAK的活性,Olumiant有望给类风湿关节炎患者带来福音。
16 Epidioloex (cannabidiol)
6月25日,美国FDA批准了GW RESEARCH LTD新药Epidiolex口服溶液治疗两岁及以上患者的两种罕见和严重癫痫,Lennox-Gastaut综合征和Dravet综合征相关的癫痫发作。这是FDA批准的第一种含有从大麻中提取的纯化药物的药物。这也是FDA首次批准用于治疗Dravet综合征患者的药物。
在三项随机,双盲,安慰剂对照的临床试验中研究了Epidiolex的有效性,该试验涉及516名患有Lennox-Gastaut综合征或Dravet综合征的患者。与安慰剂相比,Epidiolex与其他药物一起被证明可有效降低癫痫发作的频率。
17 Zemdri (plazomicin)
6月25日,美国FDA批准了Achaogen新药Zemdri上市,用于由某些肠杆菌科细菌感染引起的、治疗选择非常有限或无治疗选择的复杂性尿路感染(cUTI,包括肾盂肾炎)成人患者,该药是一种静脉输注药物,每天给药一次。
此次批准,使Zemdri成为治疗cUTI的唯一一种每日一次的氨基糖苷类疗法。plazomicin是一种新一代的氨基糖苷类抗生素,能够抑制细菌蛋白质的翻译过程。plazomicin是在西梭霉素(sisomicin)基础上进行了化学改造而得,能避免被主要的氨基糖苷类抗生素钝化酶(AME)破坏而失去活性。plazomicin开发用于治疗MDR革兰氏阴性菌肠杆菌科细菌导致的严重感染,包括对碳青霉烯类抗生素耐药的肠杆菌。
Zemdri的获批,是基于III期临床研究EPIC的数据。该研究是首个评估每日一次氨基糖苷类疗法治疗cUTI(包括肾盂肾炎)的随机对照研究,数据显示,Zemdri达到了与美罗培兰(meropenem)的非劣效性。
参考资料:
1. Novel Drug Approvals for 2018
2. Novel Drug Approvals for 2017
3.2017年FDA批准的46个新药汇总及国内进展简述(https://news.yaozh.com/archive/21707.html)
4.药智数据中国临床试验数据库
5.新浪医药新闻(http://med.sina.com/)(转化医学网360zhyx.com)
还没有人评论,赶快抢个沙发