 腾讯登录
腾讯登录【CELL DEATH DIS】南方科技大学:前沿研究!可抑制肝细胞癌的致瘤表型
| 导读 | 在包括肝细胞癌(HCC)在内的多种癌症中,肿瘤干细胞(CSCs)促进了肿瘤的发生、发展和复发。CSCs的表观遗传重编程已成为诱导从恶性到良性转变的一种有前途的策略。 |
具有PHD和无名指结构域1 (UHRF1)的泛素样蛋白是DNA甲基化遗传所必需的。在此,我们研究了UHRF1在调节CSC性质中的作用和机制,并评估了UHRF1靶向对HCC的影响。在二乙基亚硝胺(DEN)/ ccl4诱导和myc转基因肝癌小鼠模型中,肝细胞特异性Uhrf1敲除(UHRF1HKO)强烈抑制肿瘤的发生和CSC的自我更新。在人HCC细胞系中消融UHRF1产生一致的表型。整合RNA-seq和全基因组亚硫酸氢盐测序揭示了UHRF1沉默表观遗传重编程癌细胞诱导的广泛的低甲基化,使其分化和肿瘤抑制。在机制上,UHRF1缺陷上调了CEBPA,随后抑制了GLI1和Hedgehog信号传导。在myc驱动的HCC小鼠中,给药一种潜在的UHRF1抑制剂——桧木醇可以显著降低肿瘤生长和CSC表型。具有病理生理意义的是,UHRF1、GLI1和关键轴蛋白在小鼠和HCC患者肝脏中的表达水平持续升高。这些发现强调了UHRF1在肝CSCs中的调控机制,并对HCC治疗策略的发展具有重要意义。

https://www.nature.com/articles/s41419-023-05895-w
研究背景
01
肝细胞癌(HCC)占原发性肝癌病例的75-85%。HCC相关死亡率高的部分原因是晚期HCC患者比例高且缺乏有效治疗。因此,迫切需要新的治疗策略。
肿瘤干细胞(CSC)或肿瘤启动细胞模型表明,肿瘤内具有自我更新和分化特征的干细胞样细胞亚群负责肿瘤的启动,治疗抵抗和复发。这些罕见的细胞已经在包括HCC在内的各种癌症类型中被报道,并且已经鉴定出几种CSC标记物,如CD44和CD133。肝CSC表现出Wnt/β-catenin、Hedgehog和Notch信号通路的频繁激活,这些信号通路在肝脏发育、肝脏生长和肝CSC自我更新中发挥重要作用。此外,SOX2、c-MYC等与干细胞相关的转录因子在肝癌中也有异常表达。与正常组织干细胞类似,高恶性的CSCs可以转化为低致瘤性的分化细胞。CSC的这种可塑性可以通过分化治疗来消耗CSC亚群和根除癌症。
现有证据表明,DNA甲基化重编程是一种关键的表观遗传机制,在CSC可塑性中起着至关重要的作用。DNA甲基化是由DNA甲基转移酶DNMT3a和DNMT3b诱导的。在细胞分裂过程中,具有PHD和无名指结构域1的泛素样蛋白(UHRF1)将DNA甲基转移酶1 (DNMT1)招募到半甲基化的DNA位点,并维持DNA甲基化,从而使子细胞继承DNA甲基化模式。既往研究报道,消融UHRF1可导致不同成体干细胞分化。最近,Hu等报道靶向UHRF1可根除髓系白血病中的白血病起始细胞,揭示了UHRF1在维持癌起始细胞中的作用。UHRF1在几种类型的癌症中经常过表达,并在癌症进展中起致瘤作用。然而,UHRF1在肝脏中调节CSC特性的功能作用和机制尚不清楚。虽然已经有一些关于UHRF1在肝损伤和癌症中的研究,但大多数结果都是在体外的HCC细胞中获得的。对于模式生物的体内数据,有些发现似乎不一致甚至相互矛盾。例如,在斑马鱼肝细胞中,UHRF1过表达会通过破坏Dnmt1的稳定和离域导致DNA低甲基化,而Magnani等人发现,UHRF1的缺失也会导致斑马鱼肝脏中DNA甲基化丢失。此外,uhrf1缺失引起的DNA低甲基化诱导了斑马鱼转座元件的激活和干扰素反应,但在UHRF1缺失小鼠中,通过H3K27me3的重新分配,转座元件被抑制。因此,有必要在碱基分辨率上描述UHRF1耗竭时的甲基组,并研究由此产生的转录重编程。此外,还需要体内模型和数据来确定UHRF1是否可以作为HCC的治疗靶点。
在这里,我们证明了UHRF1作为一种表观遗传调节剂,通过GLI1/Hedgehog和Wnt信号重编程CSCs向分化和肿瘤抑制方向发展。此外,通过基因敲除或药物抑制来消融UHRF1可减轻小鼠的肝癌发生和CSC表型。我们的研究结果提供了机制见解,并确定了UHRF1作为肝癌治疗的潜在靶点。
研究过程
02
临床样本
在知情同意的情况下,从中山大学肿瘤中心(中国广州)接受肝切除术的患者中收集原发性HCC标本和配对的非肿瘤组织。采用75对冷冻原发肿瘤及邻近非肿瘤组织(队列1)反转录定量PCR (RT-qPCR)分析mRNA表达,8对western blotting (WB)分析蛋白表达。采用组织微阵列(TMA)包含177个原发性HCC肿瘤组织(队列2)用于免疫组化检测蛋白表达。本研究使用的临床标本经中山大学癌症中心人体受试者研究伦理审查委员会批准。
动物实验
所有动物实验均经南方科技大学机构动物爱护与利用委员会审核通过。所有小鼠均具有C57bl/6基因背景。通过将UHRF1flox/flox小鼠(上海模式生物中心)与白蛋白cre (Alb-Cre)小鼠(上海模式生物中心)杂交获得肝细胞特异性UHRF1敲除(UHRF1HKO)小鼠。得到的UHRF1flox/floxAlb-Cre小鼠为实验组(n = 6), UHRF1flox/flox小鼠为对照组(n = 5)。在这项研究中只使用了雄性小鼠。
为了建立纤维化相关性HCC模型,在出生两周内腹腔注射20 mg/kg DEN (Sigma, # 0258-1g)以初始化HCC过程。然后,四氯化碳(CCl4)(5µl/g体重,用橄榄油稀释)在间隔6周后每周腹腔注射两次,以促进HCC的进展。从两组小鼠身上采集组织。第一组为5只对照小鼠,7个月时处死6只Uhrf1HKOmice,用于评价敲除UHRF1对HCC癌变的影响。第二组分别于4、5、6、7月龄处死3只对照组小鼠和3只UHRF1HKO小鼠。该队列用于跟踪UHRF1在HCC癌变不同阶段的表达。收集肝组织用于后续实验。
为了建立Myc驱动的HCC模型,将hip11 -stopflox/flox-Myc小鼠(上海模式生物中心)与白蛋白cre小鼠杂交,产生肝细胞特异性Myc敲入小鼠(MycHKI/+)。通过将UHRF1flox/floxAlbCre/+和UHRF1flox/floxMycHKI/+小鼠杂交产生肝脏特异性UHRF1基因敲除小鼠(UHRF1HKOMycHKI/+)。UHRF1HKOMycHKI/+小鼠为实验组(n = 6), MycHKI/+小鼠为对照组(n = 6)。9周龄处死小鼠,收集肝组织用于后续实验。
对于扁桃酚治疗,MycHKI/+小鼠从出生4 ~ 9周开始,每周2次腹腔注射载体(对照组,n = 6)或25 mg/kg扁桃酚(治疗组,n = 8)。9周龄处死小鼠,收集肝组织用于后续实验。
统计数据
采用SPSS和GraphPad Prism进行统计分析。使用非配对学生t检验来检查任何两个预选组之间的差异。生存率差异采用Kaplan-Meier曲线和log-rank检验进行分析。采用Spearman相关分析分析两个统计变量之间的相关性。结果表示为平均值±平均值的标准误差。P < 0.05为差异有统计学意义。
研究结果
03
肝细胞特异性敲除UHRF1可减轻DEN/ ccl4诱导的肝癌发生
为了研究UHRF1功能丧失对HCC发生和进展的影响,我们通过将UHRF1flox/flox小鼠与白蛋白cre小鼠杂交,产生了肝细胞特异性UHRF1敲除小鼠(UhRF1HKO)。UHRF1HKO小鼠正常发育为可存活的成年小鼠,在体重、肝脏大体外观或组织学评估的肝脏结构方面与年龄匹配的对照组小鼠没有差异。通过单次注射DEN然后重复给药CCl4诱导纤维化相关小鼠肝癌发生,100%的小鼠在7月龄时发生肝脏肿瘤(图1A和S1A)。在UHRF1HKO肝脏中,UHRF1 mRNA表达(图S1B)和蛋白水平(图1B)均显著降低,证明了这种敲除策略的有效性。敲除Uhrf1显著降低了肿瘤的发生率和体积,而对照小鼠的肝脏则被多个肿瘤占据(图1A和S1A)。苏木精和伊红(H&E)染色证实了相应肝组织的组织学结构(图1C)。在UHRF1HKO小鼠中检测到肿瘤数量、肝脏重量和肝脏体重比明显下降,其中三只没有形成肿瘤(图1D)。DEN/ ccl4诱导HCC的转录组学分析显示,UHRF1敲除后958个基因下调(图S1C),其中Wnt信号通路富集(图1E和S1E)。共有551个基因在分子分解代谢和代谢过程中被上调和富集(图S1C和D)。此外,UHRF1的缺失降低了已知CSC特征基因(Cd24、Cd44、Cd133、Epcam、Krt19、Afp、Anpep、Icam1、Foxm1、Dlk1、Gpc3、Mycn、Sox9和Hnf4a)的表达(图1F),其中Foxm1被报道促进了UHRF1的表达[23]。相反,UHRF1的缺失增强了与肝细胞分化相关的基因(E2f7、E2f8、Cps1和Pck1)的表达(图1F)。这些体内结果表明,UHRF1基因敲除可能通过调节肝CSCs的自我更新和分化,强烈地减弱了肝癌的发生。
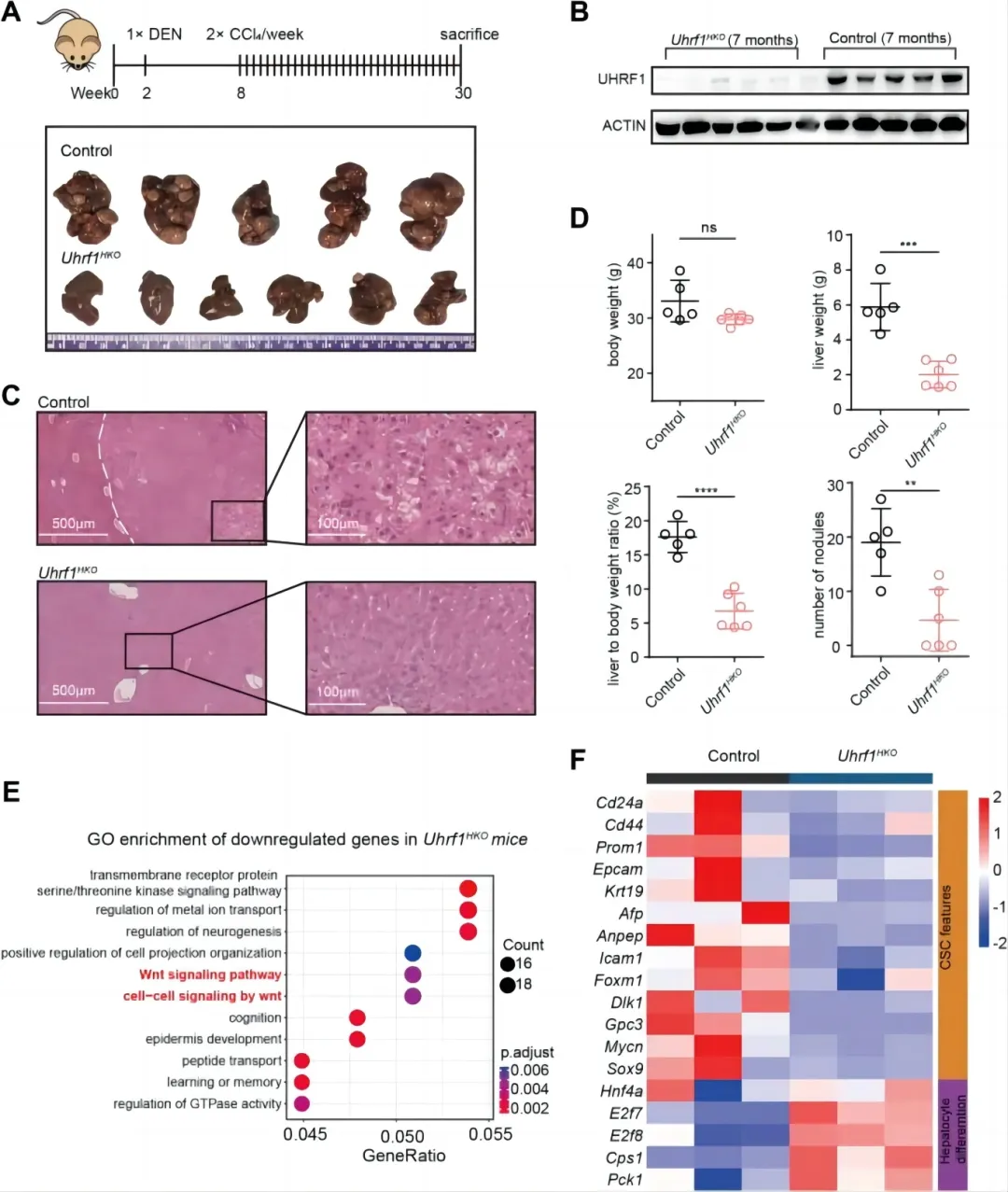
肝细胞特异性敲除Uhrf1可减轻DEN/ ccl4诱导的肝癌发生
UHRF1是维持CSC表型所必需的
先前的研究已经确定了癌症中干细胞和分化的基因表达特征。Yamashita等人发现,在肝干细胞样hcc中,一个基因簇表达上调,一个基因簇表达下调,与成熟肝细胞功能相关。Rhodes等人描述了在各种类型的未分化癌症中通常被激活的转录谱[24,25]。基因集富集分析显示,上述两个干细胞相关基因集在UHRF1 mRNA高表达的肿瘤中呈正富集,而在TCGA-LIHC数据集中的UHRF1 mRNA高表达的患者中,分化相关基因集呈负富集(图2A)。此外,UHRF1的表达与典型Hedgehog和Wnt信号通路的激活呈正相关(图S2A)。我们之前建立了体外肝细胞分化模型[26],发现UHRF1在肝脏发育和肿瘤发生过程中表现出类似癌胚的基因表达模式(图2B)。这些发现表明,UHRF1调节肿瘤起始细胞的干性。支持这一观点的是,在小鼠模型中,UHRF1的表达水平在HCC发病过程中逐渐升高,同时CD133和CD44蛋白水平升高,而UHRF1的缺失几乎完全消除了这种升高(图2C、D和S2B)。为了进一步研究UHRF1是否影响人肝脏CSC属性,我们在SNU-449和CRL-8024 HCC细胞系中建立了稳定的UHRF1敲低或过表达(图S2C和S3A)。UHRF1的异位过表达并不影响肿瘤细胞的增殖或迁移,这可能是由于功能冗余(图S3B-D)。相比之下,UHRF1沉默显著降低了致瘤性和迁移(图S2D-F)。球体形成实验显示,UHRF1沉默显著减少了干细胞形成的球体数量(图2E)。流式细胞术证实,UHRF1敲除细胞中CD44+CD133+肝CSCs的比例低于对照细胞(图2F)。同样,在UHRF1耗尽后,CD133和CD44的免疫荧光信号显著降低(图2G)。总的来说,这些结果表明UHRF1在CSC维持中起关键作用。(转化医学网360zhyx.com)
参考资料:
https://www.nature.com/articles/s41419-023-05895-w
注:本文旨在介绍医学研究进展,不能作为治疗方案参考。如需获得健康指导,请至正规医院就诊。










还没有人评论,赶快抢个沙发