 腾讯登录
腾讯登录【Nature子刊】军事医学科学院伯晓晨团队开发新一代深度学习框架,可预测癌症基因模块
| 导读 | 在本研究中,团队开发了CGMega,这是一个可解释的基于图注意力的深度学习框架,用于执行癌症基因模块解剖。 |
2024年7月17日,军事医学科学院伯晓晨团队在期刊《Nature Communications》上发表了题为“CGMega: explainable graph neural network framework with attention mechanisms for cancer gene module dissection”的研究论文。研究结果表明,CGMega可用于剖析癌症基因模块,并为癌症发展和异质性,提供高阶机制和见解。

https://www.nature.com/articles/s41467-024-50426-6
研究背景
01
活细胞的复杂功能,是通过许多基因和基因产物的协同活动进行的。细胞的大部分活动,被组织成基因模块:一组共同调节,以响应不同条件的基因。主动驱动模块可以触发癌症的标志,并赋予癌细胞适应性优势。癌症基因模块的阐明,可以大大加深科学界对癌症发展的理解,并为最佳治疗方法的设计,提供信息。
自高通量测序技术发展以来,基因模块检测方法,一直是大型基因药典生物学解释的基石。目前使用的方法,主要有两个缺点。(1)最近的染色体构象捕获技术,揭示了三维(3D)基因组结构,并证明了其在建立基因-基因关系方面的关键作用。(2)基因模块表现出高阶网络特征,高阶相互作用,调控生物系统中的复杂功能。 现行大多数共表达聚类或基于相关性的方法,都无法将重要的组学特征,分配给模块基因。
图神经网络(GNN)是测量图结构数据(如生物网络)的有力方法,并成功对PPI、Hi-C数据,以及跨蜂窝网络发现基因模块,进行建模。GNN能够处理不同的Hi-C表示,即将基因属性作为节点特征,或将基因之间的关系,作为图边。团队已经证明GNN及其解释技术,是剖析基因组相互作用之间高阶关系的有力工具。
在本研究中,团队提出了一个新框架(CGMega),用于剖析具有可解释图注意力的癌症基因模块。团队将CGMega应用于乳腺癌细胞系和急性髓系白血病(AML)患者,并揭示了癌症基因模块中基因之间的高阶关系。CGMega共同利用了最近GAT在多组学数据上的出现,并获得了对癌症基因模块层次结构的基本发现和理解。
研究进展
02
CGMega在癌症基因预测中有效
CGMega基于癌症基因的准确预测,鉴定了基因模块,因此,团队测试了CGMega在MCF7细胞系上癌症基因预测的性能。MCF7细胞系是一种具有高置信度多组学数据的人乳腺癌细胞系。CGMega实现了0.9140AUPRC和0.9630的受试者工作特征曲线下面积(AUROC)。为了证明CGMega在癌症基因预测任务中的进展,团队将CGMega与各种方法进行了比较,包括通用模型GCN、GAT、MLP、SVM,以及为癌症基因分类设计的特定模型,包括MTGCN42、EMOGI25和 MODIG43。通过计算AUPRC、AUROC、准确度(ACC)和F1分数,CGMega在这四个指标上的表现,优于所有其他方法。
团队采用了CGMega的两步法。在初始阶段,CGMega在MCF7细胞系上进行了预训练,使其能够掌握癌症基因中普遍存在的基本模式和特征。在预训练之后,团队对其他癌症进行了微调,使CGMega能够适应和微调其学习的表征,以适应这些罕见癌症的特定环境。
为了评估迁移学习的性能,团队使用K562细胞系上的所有标记基因(597个阳性和1,839个阴性)对非预训练的CGMega(从头开始训练)和预训练的CGMega进行了测试。随着标记基因数量的减少,非预训练的CGMega的性能急剧下降,而预训练的CGMega继续保持高性能。此外,Hi-C特征在预测方面表现出强大的改进,特别是当标记的基因小于200时。团队比较了CGMega与其他方法中小样本迁移学习的性能,预训练的CGMega具有最高值。
CGMega利用15维基因特征,包括10维组学特征和5维浓缩Hi-C特征,这些特征源自Hi-C数据的降维。团队观察到,组学和Hi-C特征,都对模型预测做出了贡献。此外,具有5维浓缩Hi-C特征的CGMega,不如具有10维组学特征的CGMega,表明结构特征可能对组学特征的质量具有补偿作用。
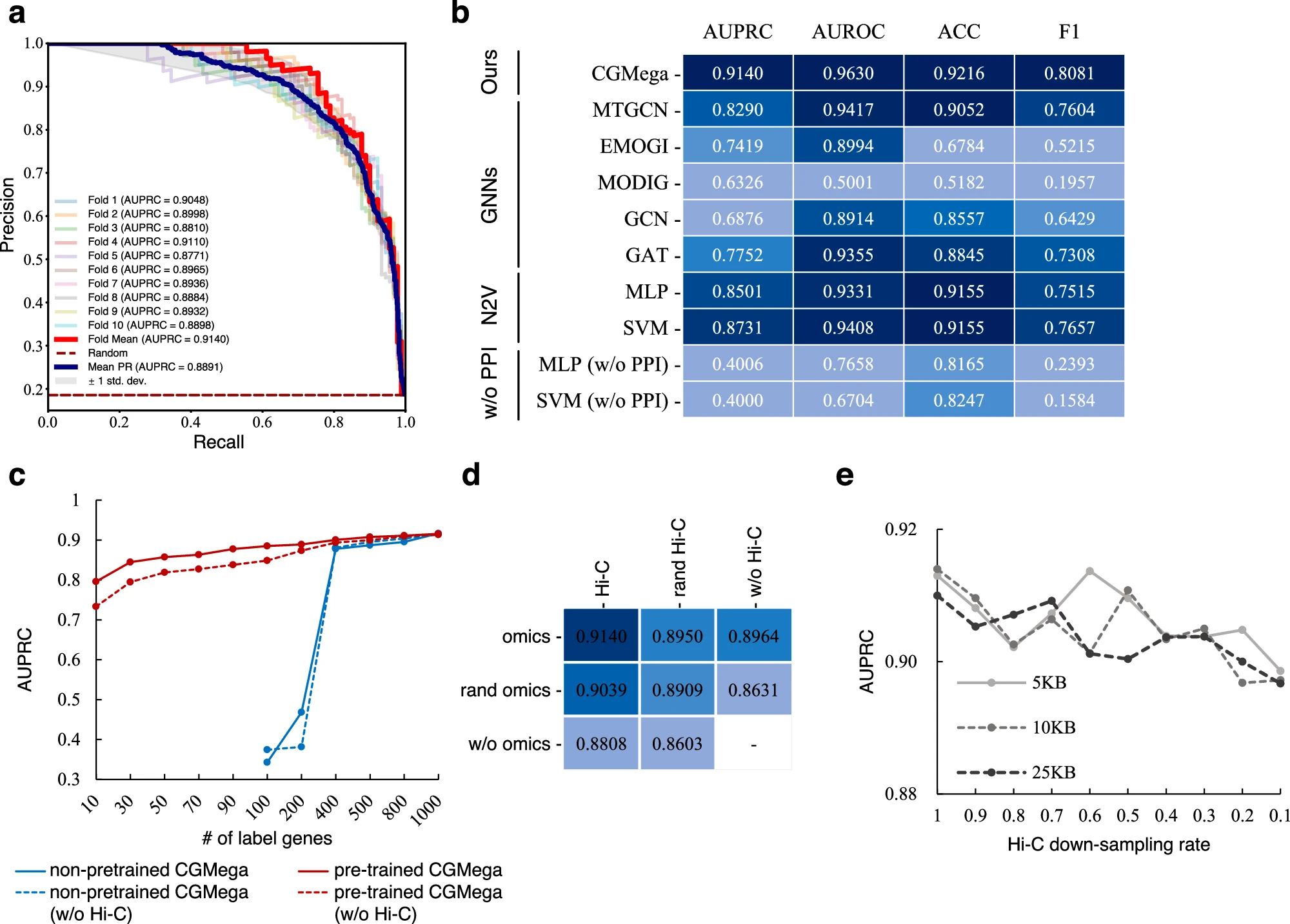
CGMega在癌症基因预测任务中的表现。
CGMega为多组学数据整合提供新策略
CGMega的卓越表现,得益于多组学信息的有效整合,包括基因组、表观基因组、PPI,尤其是3D基因组架构。Hi-C是目前用于研究3D基因组组织的最广泛使用的检测方法。然而,将Hi-C数据与其他组学数据一起测量,通常受到其噪声、稀疏性和可变分辨率的限制。为了在癌症基因预测任务中获得最佳性能,团队测试了具有不同Hi-C数据嵌入的集成方法。
通过系统地比较不同的集成方法与Hi-C数据嵌入,团队发现,在癌症基因预测任务中,使用Hi-C潜在特征作为基因特征,优于直接测量Hi-C数据作为基因相互作用。SVD是一种有效的降维方法,用于将Hi-C数据与其他组学数据相结合。
人乳腺癌细胞系中具有多组学特征的基因模块
CGMega基于与模型无关的神经网络解释方法,检测基因模块。团队将CGMega应用于人类乳腺癌MCF7细胞系,并检查了358个已知癌症基因的模块。这些癌症基因并非随机分散在基因模块中,它们往往位于同一模块中。在这些基因模块中,TP53的富集程度最高,参与了139个癌症基因模块,其次是ESR1(63个参与)和AKT1(61个参与)。除了这些众所周知的癌症基因外,团队还观察到另外12个高度参与模块的基因,例如XPO1、NCOR2和PPM1A。团队还研究了基因模块的图形指标的结构特征,包括传递性、聚类系数、度中心性和介介中心性。癌症基因模块的拓扑结构,明显优于非癌症基因模块(P< 2.47e-5,配对t检验)。
除了基因模块的拓扑结构之外,团队还研究了特征重要性得分。CGMega利用15维多组学特征作为输入,并为每个特征,生成重要性分数。团队从TCGA项目中,收集了乳腺癌的RNA-seq数据,并鉴定了差异表达基因。DEGs的比例,在簇-3中最高。根据CGMega预测,Hi-C与其他活性调控元件一起,对这些基因具有共同作用。
基于特征重要性得分,团队提出代表性特征(RFs),作为重要性得分排名靠前的特征。团队重点关注BRCA1和BRCA2的基因模块,这是乳腺癌中最常见的基因。团队观察到,它们的基因模块之间的拓扑差异。简而言之,BRCA1是一种在DDR的多个阶段起作用的多效性DNA损伤反应(DDR)蛋白,也被发现与另外20个基因广泛相关。相比之下,BRCA2作为同源重组(HR)核心机制的介质,通过直接介导HR修复的重要基因ROCK2,与其他基因连接。基于TCGA项目的基因表达数据,团队发现,ROCK2在乳腺肿瘤供体中,与BRCA2表达呈正相关,而在正常乳腺组织中,没有这种相关性。BRCA2和ROCK2在乳腺癌中的共表达,表明BRCA2抑制剂在肿瘤发生中的联合作用,这可能指导BRCA2抑制剂对肿瘤细胞的作用增强。研究结果表明,BRCA2和ROCK2抑制剂联合治疗24小时后抑制MCF7肿瘤细胞,比单独使用BRCA2抑制剂更有效。这是一种增强BRCA2抑制剂敏感性的潜在策略。此外,SNV是BRCA1和BRCA2的RF。团队还观察到一个由BRCA1基因模块和BRCA2基因模块通过TP53、SMAD3和XPO1三个共享基因组合的高阶基因模块。综上所述,这些适应症,意味着CGMega能够检测具有多组学特征的可解释和高阶基因模块。
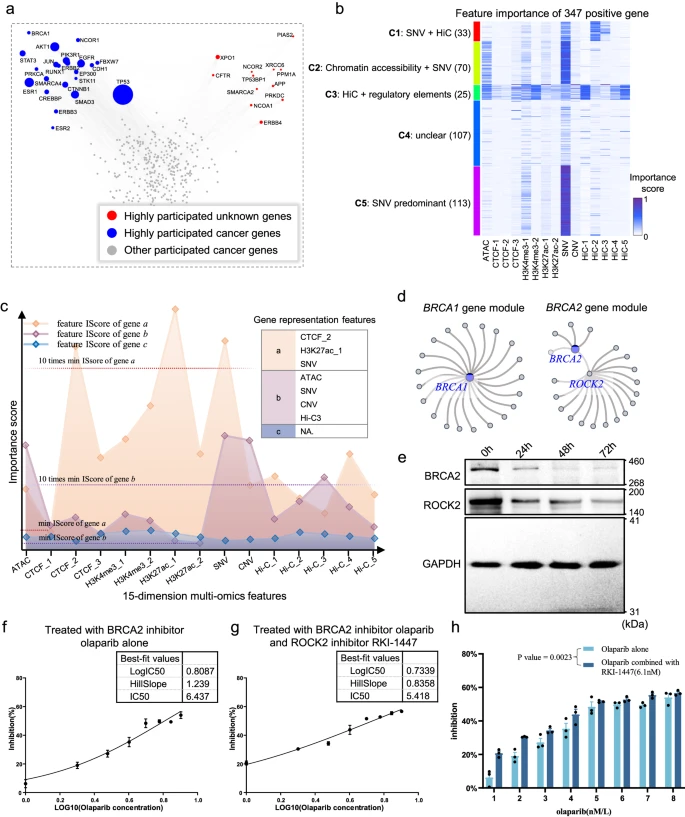
乳腺癌细胞系中的基因模块。
研究结论
03
CGMega与其他方法的主要区别在于:(1)与现有方法相比,CGMega在捕获3D基因组结构方面,具有先进的能力,这已被广泛证明,是癌症研究的新视角。(2)CGMega使用GNNExplainer40,解释癌症基因预测的促成因素。(3)CGMega显示了不同癌症之间的知识可转移性。研究结果证明了,CGMega在不同癌症类型上的可转移性,这是本研究的一个重要方面。
除了CGMega的这些优点外,团队还对Hi-C数据与其他组学数据的整合方法,进行了全面评估,并证明了:(1)图结构在整合多组学信息方面是先进的,特别是对于分子信号和基因关系组合;(2)使用SVD将Hi-C数据编码为基因特征,优于将Hi-C数据作为基因连锁进行测量。
CGMega在乳腺癌细胞系和AML患者中的应用,有助于发现:(1)癌症基因模块广泛且组织良好,包括以癌症基因为中心的模式和非癌症基因中心模式;(2)癌症基因(已知的癌症基因和预测的癌症基因)往往富集在一个模块中,表明癌症基因在肿瘤发生中,具有复杂的相互作用;(3)除了这些众所周知的癌症基因外,还有一些枢纽基因位于癌症基因模块的中心,或存在于数十个癌症基因模块中。此外,CGMega在乳腺细胞系(AUPRC = 0.9140)和AML患者(平均AUPRC=0.8528)中的良好表现表明:(1) CGMega对细胞系和供体样本,以及实体瘤和液体肿瘤研究均表现出疗效;(2)CGMega对于缺少分子特征的输入是灵活的。这表明,团队的框架可能适用于其他类似的任务。
参考资料:
1.Segal, E. et al. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 34, 166–176 (2003).
2.Kamimoto, K. et al. Dissecting cell identity via network inference and in silico gene perturbation. Nature 614, 742–751 (2023).
还没有人评论,赶快抢个沙发