 腾讯登录
腾讯登录复旦大学附属中山医院蓝斐团队:揭秘SMYD5—核糖体甲基转移酶如何加速肝细胞癌发展!
| 导读 | 在本研究中,团队报告了SMYD5对RPL40 K22具有强大的体外活性,并且主要在细胞中催化RPL40 K22me3。 |
2024年8月5日,复旦大学附属中山医院蓝斐团队在期刊《Cell Research》上发表了题为“SMYD5 is a ribosomal methyltransferase that catalyzes RPL40 lysine methylation to enhance translation output and promote hepatocellular carcinoma”的研究论文。在本研究中,团队将SMYD5鉴定为负责RPL40 K22me3的核糖体蛋白甲基转移酶,随后评估这种修饰在翻译中的作用。团队证明RPL40 K22上的SMYD5甲基化增强了全局翻译输出,并且是正确翻译延伸所必需的。

https://www.nature.com/articles/s41392-024-01885-2#Sec4
研究背景
01
蛋白质赖氨酸Nε-甲基化在各种生物过程中,起着至关重要的作用。虽然在过去20年左右的时间里,它对组蛋白转录调控的影响已经得到了广泛的研究,但它在翻译中的作用,在很大程度上仍未得到探索。RPL40是一种由UBA52基因编码的特殊核糖体蛋白。前体UBA52蛋白是一种由128个氨基酸(aa)组成的融合蛋白,包含泛素模块(76 aa)的N端融合。去除泛素后,RPL40的成熟形式长度为52 aa,是细胞质中最后组装成60S核糖体亚基的成分之一。在成熟的80S核糖体中,RPL40位于P柄/GTP酶激活中心(GAC)和肌肽-蓖麻毒素环(SRL)附近,其中延伸因子eEF1A和eEF2结合。延伸因子对于将肽基tRNA募集到A位点,以及将其从A位点转移到P位点,至关重要。RPL40已被提议选择性地调节应激相关的mRNA翻译,并赋予酵母对伸长抑制剂Sordarin的抵抗力,表明RPL40在蛋白质合成中,具有重要功能。
SET和含MYND结构域(SMYD)蛋白,构成了赖氨酸甲基转移酶进化上保守的亚家族,其特征在于催化SET结构域被MYND结构域分裂。其中,SMYD5可催化病毒Tat蛋白甲基化,参与HIV感染,以及在启动子上催化组蛋白H3K36me3,并驱动肝细胞癌(HCC)的肿瘤发生,尽管下游机制尚不清楚。事实上,大多数癌症类型的SMYD5 mRNA水平都升高,其中HCC是最重要的类型之一。最近的两项多组学研究发现,HCC样本中SMYD5 mRNA和蛋白质水平均显著升高,并且与不良临床结果相关。
在本研究中,团队将SMYD5确定为一种核糖体赖氨酸甲基转移酶,主要催化RPL40 K22me3。SMYD5-RPL40 K22me3轴对于高效的翻译伸长和整体蛋白质合成,至关重要。HCC癌细胞中SMYD5的缺乏,导致对mTOR抑制剂的超敏反应,这可能是由于对蛋白质合成的复合抑制作用。采用离体和体内HCC模型,团队进一步阐明了SMYD5介导的RPL40 K22me3在维持癌症生长中的关键作用,尤其是在mTOR信号传导受到抑制的情况下。这些发现强调了靶向SMYD5-RPL40 K22me3轴,作为HCC患者治疗策略的潜力。
研究进展
02
SMYD5缺失使癌细胞对mTOR通路阻断敏感
与对照细胞相比,导致SMYD5 KO细胞适应性降低的4个主要标志物是mTOR通路的抑制剂,包括雷帕霉素、Torin1、Omipalisib(双重mTOR/PI3K抑制剂)和抑制 4EBP1磷酸化的凋亡诱导剂星形孢菌素。在SMYD5丢失时,Huh7细胞对mTOR抑制剂、Torin1和雷帕霉素的敏感性升高,使用IC50值偏移了大约5倍。实验表明,与WT RPL40相比,在Torin1处理下,K22R突变在恢复Huh7生长潜力方面存在缺陷。在SNU449、HepG2和HeLa细胞中,还观察到SMYD5丢失后,对mTOR抑制的敏感性升高。
与基线条件相比,在mTOR抑制下,Huh7和SNU449细胞系中新合成的蛋白质的SMYD5依赖性减少更明显。在293T RPL40 K22R KI细胞系中,观察到新合成蛋白的减少增加。SMYD5丢失后,有大量mRNA在SMYD5丢失后TE减少(可靠检测到的总数9,496个中的491个,FC> 2,P调整<0.05),而没有mRNA显示TE显著增加。当临界值降低到FC 1.5时,2,451个mRNA的TE降低,而没有mRNA显示,TE显著增加 。在SMYD5 KO细胞中,在Torin1处理下的CDS区域,观察到核糖体结合的全局减少更大。同样,在Torin1处理下,未检测到差异密码子占用率,与未处理的正常情况相同。
与Torin1处理的SMYD5 KO相比,G2M检查点、MYC靶点、E2F靶点和核糖体生物发生在对照细胞中富集,符合SMYD5 KO细胞在此条件下的生长缺陷。与对照细胞相比,Huh7和HeLa SMYD5 KO细胞的40S、60S、80S和多糖体丰度均较少。在mTOR抑制后,SMYD5 KO Huh7细胞中的一些核心核糖体蛋白适度减少,表明SMYD5丢失和mTOR抑制的综合效应,可能会影响核糖体生物发生,部分解释了SMYD5耗尽细胞在mTOR抑制下的生长潜力降低。综上所述,当用Torin1处理时,SMYD5 KO细胞会经历大量的翻译抑制。
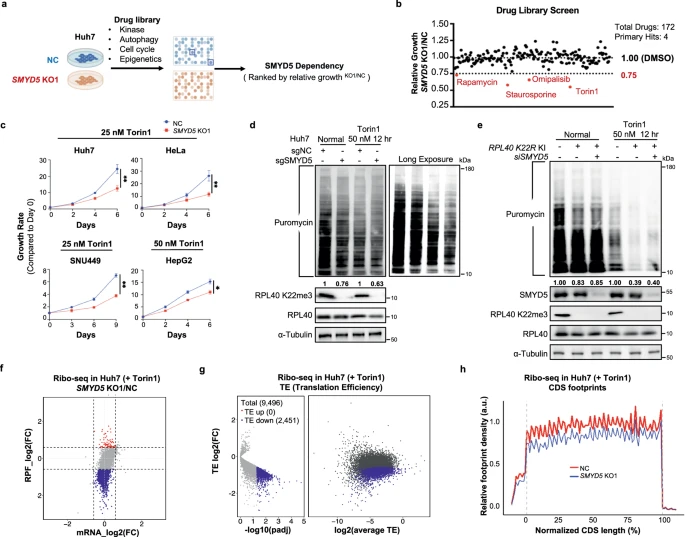
SMYD5缺失使癌细胞对mTOR阻断敏感。
SMYD5促进肝细胞癌体内发育
肝细胞癌最常作为肝硬化的并发症发生。肝脏特异性SMYD5耗竭的动物发育正常,没有任何明显的表型,与国际小鼠表型分析联盟的数据一致,该数据显示全身Smyd5 KO小鼠是可行的,可育的,并且仅表现出较小的表型。团队利用基于两阶段化学应用的模型,来启动和促进与晚期肝纤维化相关的肝细胞肿瘤。HCC模型是通过单次注射具有遗传毒性的N-亚硝基二乙胺(DEN)诱导的,然后重复给予促纤维化剂四氯化碳(CCl4)。对照动物在6个月大时,发展为晚期HCC。相比之下,SMYD5消融导致整体肿瘤大小和肿瘤负荷显著减小,观察到的肿瘤结节较少。肝脏/体重比是HCC肿瘤总负荷的常见指标,在Smyd5 KO动物中也显著降低。此外,SMYD5的耗竭导致RPL40 K22me3染色信号完全丧失和肿瘤癌细胞增殖(pH3)的衰减。

SMYD5在体外和体内均促进HCC。
研究结论
03
在本研究中,团队将SMYD5鉴定为负责RPL40 K22me3的核糖体蛋白甲基转移酶,随后评估这种修饰在翻译中的作用。团队证明RPL40 K22上的SMYD5甲基化增强了全局翻译输出,并且是正确翻译延伸所必需的。SMYD5的缺失和RPL40上的甲基化,导致核糖体碰撞升高和对靶向A位点的翻译抑制剂的超敏反应。SMYD5缺失会导致数千个翻译mRNA的TE强烈减少,而不涉及密码子特异性效应 。基于这些发现,团队得出结论,SMYD5-RPL40 K22me3轴是一般伸长所必需的,但不是选择性平移所必需的。SMYD5可能会影响起始和60S组装,这需要进一步的研究。
SMYD5和RPL40 K22me3轴在低水平伸长应力条件下增强了核糖体适应性,特别是与A位点扰动相关。该轴是否是核糖体应激感应途径的一个组成部分,是未来研究的一个有趣的问题。团队没有观察到由于翻译因子结合而导致的RPL40及其K22侧链的显著改变。因此,RPL40 K22me3如何影响A位点功能和GAC/P柄动力学的详细机制尚不清楚,团队鼓励未来的研究朝这个方向发展。
RPL40在测试的人类细胞系和小鼠组织中接近完全甲基化,但未来应彻底研究RPL40 K22me3的生理/病理范围,以及SMYD5本身的调控。虽然mTOR抑制剂在治疗肝细胞癌方面的疗效有限,但是,破坏SMYD5介导的RPL40 K22me3甲基化,可增强HCC细胞对mTOR抑制的敏感性。这表明,双重靶向方法,可能超过单独使用mTOR抑制的有效性。
SMYD5耗竭单独显著抑制癌症生长,并使它们对mTOR抑制过敏。此外,肝硬化相关HCC与肝细胞特异性Smyd5 KO的基因工程小鼠模型表明,SMYD5-RPL40 K22me3通路在肿瘤发生和发展中至关重要。与其在细胞培养测定中的有限效果相比,在动物模型中观察到的SMYD5消融的稳健表型,可以通过体内mTOR活性受限来解释,这是由于营养和氧气供应有限。因此,团队建议将SMYD5作为HCC的新治疗靶点,其疗效可以与mTOR抑制相结合。由于SMYD5在其他癌症类型中也升高(TCGA),未来的研究应侧重于开发SMYD5的特异性抑制剂或调节剂,作为癌症治疗的通用方法。
靶向转化机制——该策略已在造血恶性肿瘤中取得成功,其核糖体靶向剂(Homoharringtonine,也称为Omacetaxine)由中药开发,并于2012年获得FDA的加速批准— 核糖体修饰剂仍未得到充分探索。在本研究中,团队发现靶向SMYD5可能表现出良好的安全性。与直接靶向核糖体相比,靶向核糖体修饰剂SMYD5,可能存在较少的毒性问题。
与组蛋白赖氨酸甲基化在转录调控中公认的作用相反,核糖体蛋白中赖氨酸甲基化在翻译过程中的功能,在很大程度上仍未得到探索。与核小体类似,核糖体是由带正电荷的小核糖体蛋白制成的核糖核蛋白复合物,富含赖氨酸和精氨酸,以及带负电荷的rRNA。这种物理化学特性为类似的表观遗传机制的存在和直接影响核糖体,奠定了基础。目前尚不清楚赖氨酸甲基化是否发生在更多的核糖体蛋白上,特别是那些位于当前结构分析无法看到的灵活调控区域的核糖体蛋白。因此,团队呼吁在这个新兴领域进行未来的研究,旨在将对赖氨酸甲基化重要性的理解扩展到转录之外,包括核糖体生物学。
参考资料:
1.Williamson, N. A., Raliegh, J., Morrice, N. A. & Wettenhall, R. E. Post-translational processing of rat ribosomal proteins. Ubiquitous methylation of Lys22 within the zinc-finger motif of RL40 (carboxy-terminal extension protein 52) and tissue-specific methylation of Lys4 in RL29. Eur. J. Biochem. 246, 786–793 (1997).
2.Natchiar, S. K., Myasnikov, A. G., Kratzat, H., Hazemann, I. & Klaholz, B. P. Visualization of chemical modifications in the human 80S ribosome structure. Nature 551, 472–477 (2017).










还没有人评论,赶快抢个沙发