 腾讯登录
腾讯登录诱导细胞焦亡!中山大学李轩教授团队:揭示肿瘤联合治疗新策略
| 导读 | 在许多肿瘤中,泛素连接酶MDM2的高表达是p53失活的主要原因,使其成为一个有希望的治疗靶点。然而,MDM2抑制剂在临床试验中失败,因为p53诱导的反馈增强了MDM2的表达。因此迫切需要找到一种有效的适应性基因型或靶标组合。 |
8月30日,中山大学李轩教授团队在期刊《Journal of Experimental & Clinical Cancer Research》上发表了研究论文,题为“Synthetic lethality of combined ULK1 defection and p53 restoration induce pyroptosis by directly upregulating GSDME transcription and cleavage activation through ROS/NLRP3 signaling”,本研究中,研究人员通过高通量筛选,确定ULK1是MDM2抑制剂APG115的合成致死基因。结果表明,ULK1的缺失显著增加了敏感性,细胞发生典型的焦亡。在机制上,p53通过直接介导GSDME转录诱导基础水平的焦亡,从而促进焦亡的发生。此外,ULK1缺失减少了线粒体自噬,导致受损线粒体的积累和随后活性氧(ROS)的增加。这反过来又通过NLRP3-Caspase炎症信号轴切割和激活GSDME。另外,铂耐药肿瘤中线粒体自噬增强,ULK1耗竭/p53激活对这些肿瘤具有协同致死作用,直接通过GSDME诱导细胞焦亡。

https://jeccr.biomedcentral.com/articles/10.1186/s13046-024-03168-8#Sec24
背景知识
01
肿瘤蛋白p53 (TP53)是细胞命运的核心决定因素,通过直接转录激活调控细胞周期阻滞和程序性细胞死亡途径的激活等细胞过程。在绝大多数肿瘤中P53是无活性的。虽然野生型p53基因在肿瘤中的比例高达50%,但泛素连接酶MDM2通过蛋白酶体途径介导其泛素化及随后的降解,最终导致p53蛋白的功能缺陷。抑制MDM2活性以恢复p53功能被高度期待作为这一野生型肿瘤亚群的临床治疗策略。然而,由于MDM2的反馈激活等机制,现有的MDM2抑制剂的临床应用时间有限,且不良反应较大。临床试验的结果并不令人满意,这凸显了确定辅助治疗靶点以优化治疗策略的迫切需要。
细胞焦亡是一种新发现的不同于细胞凋亡的程序性细胞死亡方式,其发生需要炎症性半胱氨酸天冬氨酸蛋白酶(caspase)裂解和激活一类GSDM家族蛋白。GSDM裂解释放该蛋白的N末端结构域,导致细胞的裂解性死亡。GSDM家族蛋白的表达是细胞焦亡激活的关键决定因素。以往的研究从多个阶段和多个角度探讨了细胞内GSDM家族基因表达的调控,包括DNA甲基化、转录调控和蛋白质稳定性控制。这些研究结果表明,肿瘤中GSDM家族成员的失活或低表达是促进肿瘤进展和治疗抵抗的关键因素。炎症小体通路的激活是细胞焦亡的重要触发因素。然而,既往的研究主要集中在炎症小体通路在单核巨噬细胞中的作用,其是否参与上皮源性细胞的焦亡尚不清楚。
P53激活联合ULK1缺乏可启动细胞焦亡
02
研究人员进一步研究了为什么MDM2i治疗和ULK1缺乏能有效杀死癌细胞。研究人员先用APG-115预处理ULK1敲除细胞24小时,发现死亡的细胞显示出明显的细胞焦亡形态,有类似囊泡的结构从细胞膜和胞质中向细胞一极聚集。此外,通过流式细胞术检测Annexin V/碘化丙啶(PI)双阳性细胞的比例,发现在用MDM2i处理的ULK1敲除细胞中,双阳性细胞的比例显著快速增加;这种效应与在最初仅Annexin V阳性、然后逐渐变为PI阳性的凋亡细胞中观察到的效应显著不同,这表明在此条件下细胞膜的通透性发生了快速变化。此外,研究人员进行了乳酸脱氢酶(LDH)检测,观察到在用APG-115处理的ULK1敲除细胞中,LDH迅速释放到细胞外。这些结果表明,MDM2抑制剂与ULK1缺乏相结合可导致癌细胞发生强效的细胞焦亡,表现为细胞膜孔形成和细胞肿胀。为了阐明p53激活和ULK1缺乏在诱导细胞焦亡中的不同作用,研究人员在癌细胞中过表达p53或敲除ULK1,并用TNFα+cycloheximide(CHX)处理以诱导经典的细胞焦亡。为了揭示合成致死的潜在机制,研究人员检测了细胞在MDM2i/ULK1耗竭后GSDMs的mRNA表达。研究结果表明,APG-115诱导的p53激活仅显著增加了GSDME的表达水平,而对其他任何GSDM均未观察到类似效应。另外,蛋白质免疫印迹实验分析显示,ULK1敲除增强了GSDME的切割,而GSDMD的切割未显示出显著激活。总之,这些结果表明,p53和ULK1可通过不同的机制调节细胞焦亡,其中p53可能促进GSDME mRNA的转录,而ULK1缺乏则可增强GSDME蛋白的切割和激活,从而分别诱导细胞焦亡。
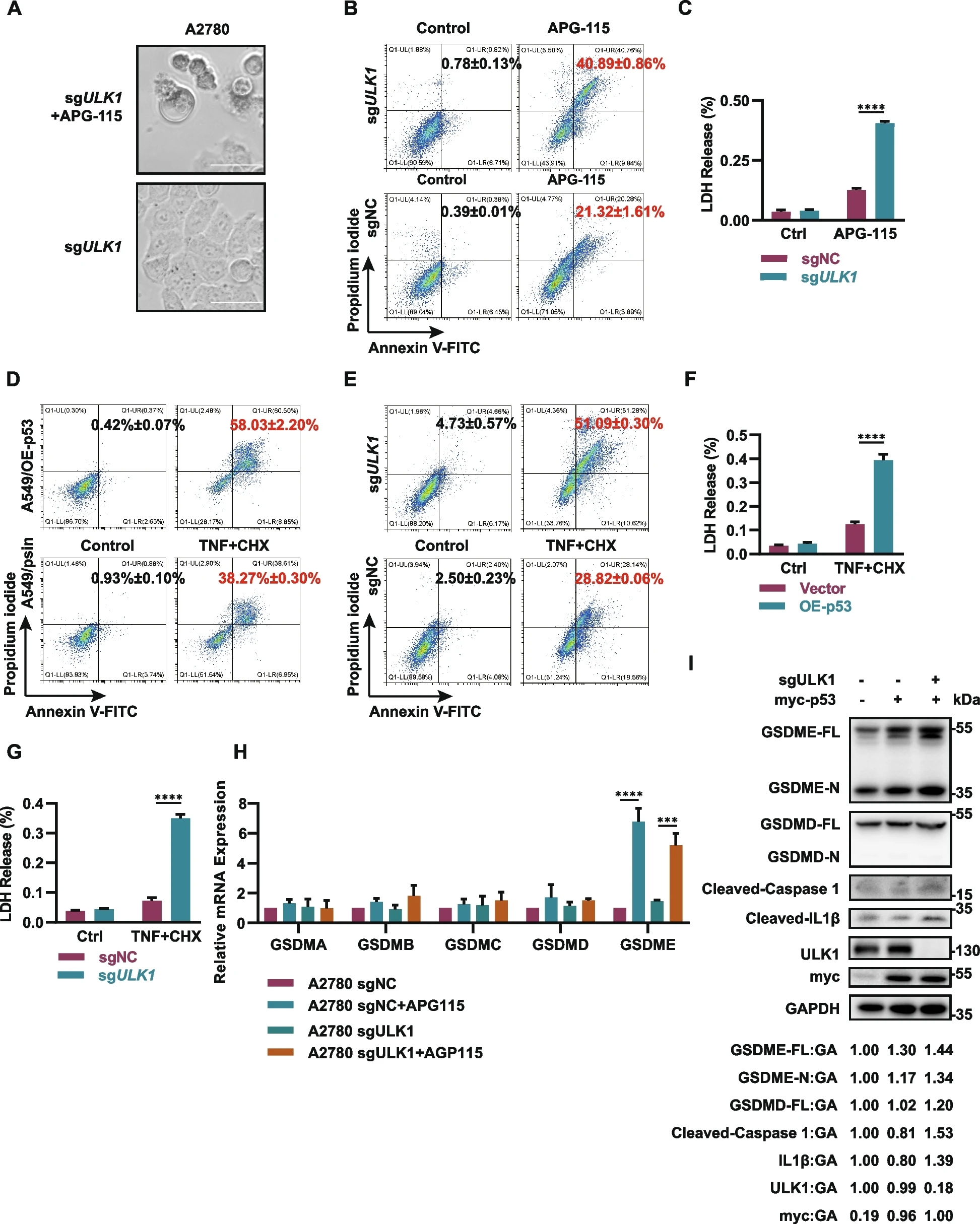
P53激活与ULK1缺乏相结合可引发细胞焦亡
线粒体自噬功能缺陷与p53激活相结合可用于逆转铂类药物耐药性
03
铂类药物通常被用作治疗实体肿瘤的主要药物,而对这种药物的耐药性是肿瘤复发和转移的主要诱因之一。研究人员已经发现,线粒体自噬在这一过程中扮演着关键角色。为了研究铂类药物耐药的机制并探索可能的治疗策略,研究人员将两个野生型p53细胞系(A549和A2780)用亚致死浓度的铂类药物处理较长时间,从而产生了对铂类药物耐药的细胞系(A549/DDP和A2780/DDP)。随后通过MTT实验确认了耐药程度。随后,通过蛋白质免疫印迹实验分析发现,耐顺铂细胞株中与自噬相关的关键基因表达水平升高,而与细胞焦亡相关的关键基因GSMDE表达水平显著降低。此外,在TNFα + CHX处理24小时后,与敏感细胞相比,耐顺铂细胞中乳酸脱氢酶的释放显著减少,Annexin V/PI双阳性信号显著降低。这些发现表明,在耐顺铂细胞株中,自噬增加,而细胞焦亡受到显著抑制。因此,可以合理地假设,靶向自噬和p53介导的细胞焦亡的联合疗法可能逆转顺铂耐药性。为了验证这一假设,研究人员采用MTT法进行了药物联合试验,结果显示,ULK1敲低联合APG-115治疗可有效诱导耐顺铂细胞死亡。该研究结果表明,线粒体自噬的激活和GSDME表达的降低可能在赋予肿瘤细胞顺铂耐药性方面发挥关键作用。此外,同时使用ULK1抑制剂和激活p53的联合治疗显示出有效消除耐药细胞的潜力,为潜在的临床干预在顺铂耐药患者中的应用提供了理论基础。
研究小结
04
总之,研究人员开发了一种创新的方法,结合MDM2抑制剂治疗,靶向与自噬相关的关键基因,以有效增强细胞焦亡,诱导肿瘤细胞死亡。这种组合疗法的协同作用及其潜在的分子机制的阐明表明,过度激活自噬的癌症对MDM2抑制剂更敏感。这一发现为激活p53的抗肿瘤疗法开辟了新的途径。此外,本研究为选择铂类耐药患者的临床治疗策略提供了重要的理论依据。(转化医学网360zhyx.com)
【参考资料】
https://jeccr.biomedcentral.com/articles/10.1186/s13046-024-03168-8#Sec24
【关于投稿】
转化医学网(360zhyx.com)是转化医学核心门户,旨在推动基础研究、临床诊疗和产业的发展,核心内容涵盖组学、检验、免疫、肿瘤、心血管、糖尿病等。如您有最新的研究内容发表,欢迎联系我们进行免费报道(公众号菜单栏-在线客服联系),我们的理念:内容创造价值,转化铸就未来!
转化医学网(360zhyx.com)发布的文章旨在介绍前沿医学研究进展,不能作为治疗方案使用;如需获得健康指导,请至正规医院就诊。










还没有人评论,赶快抢个沙发