 腾讯登录
腾讯登录安诺基因Hi-C让所有基因组组装到染色体水平
| 导读 | 目前有大量物种的基因组信息未被公布,即使已发布的基因组中,仅有26.6%的植物、12%的动物和15.4%的真菌基因组完成了染色体水平的组装;大多数基因组处于scaffold水平,这些物种需要用遗传图谱等技术将基因组组装提升至染色体水平,其中一些物种的群体构建具有很大难度,限制了基于遗传连锁图谱挂载scaffolds的可行性、精度和准确性。然而Hi-C辅助基因组组装无需遗传图谱,单个个体就能将scaffold定位到染色体上,基因组组装提升至染色体水平。 |

Hi-C技术源于染色体构象捕获技术,利用高通量测序技术,结合生物信息分析方法,研究全基因组范围内整个染色质DNA在空间位置上的关系,获得高分辨率的染色质三维结构信息。基于Hi-C数据中染色质片段间的交互强度呈现出随距离衰减的规律,Hi-C可以用于基因组组装,将杂乱的基因序列组装到染色体水平。
1、技术原理
Hi-C之所以能用于基因组组装,是基于染色质片段间的交互强度呈现出随距离衰减的规律(图1)。
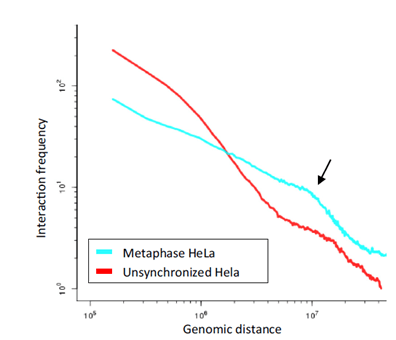
图1. Hi-C相互作用强度与距离关系[1]
其与遗传定律类似,可以用来判断scaffolds 的分类及相邻关系,分以下三步:聚类-排序-定向(cluster-order-orient),“染色体内相互作用强度高于染色体间相互作用”可以指导DNA片段的染色体分类,“同一染色体上近程相互作用强于远程相互作用”可以引导DNA片段的排序和定向。
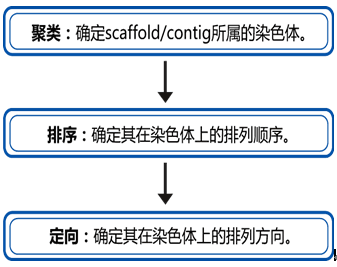
更加形象的原理展示如图2:
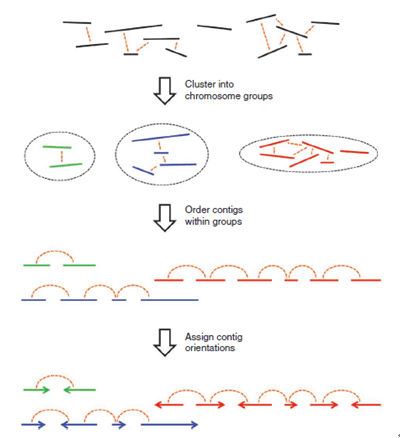
图2. Hi-C辅助基因组组装原理[2]
2. 应用范围1) 无论是Sanger测序、二代测序或者三代测序完成的,但是未组装到染色体水平的基因组。
2)基因组序列未知物种,Hi-C与常规de novo结合提高基因组组装水平。
3. 组装效果
表1 Hi-C辅助组装结果统计[2]
|
Hi-C辅助组装步骤 |
人 |
小鼠 |
果蝇 |
拟南芥 |
|
聚类的scaffold比例 |
98.2% |
99.5% |
89.5% |
84.1% |
|
排序的scaffold比例 |
99.2% |
99.5% |
94.8% |
97.3% |
|
定向scaffold比例 |
97.5% |
95.4% |
93.9% |
97.9% |
1)Hi-C判定scaffolds 的相对位置,具有更高覆盖率和特异性
Hi-C获得的reads长度是一般遗传标记的4-5 倍,使其定位具有更高的位点特异性。此外,
Hi-C能获取全基因组所有的scaffolds片段的相互作用信息,因此绝大部分scaffolds都能被组装。
2)结果更加直接、可靠
此方法基于单一株系Hi-C结果,利用scaffolds 在体内染色质相互作用的分布特征来判定染色体片段之间的邻接关系,比利用亲本后代遗传连锁交互的组装方法更为直接和可靠。
3)适用于多年生高杂合物种
对于杂合度较高的多年生物种如杨树、楸树等,群体的构建具有很大的困难,限制了基
于遗传连锁图谱挂载scaffolds的可行性、精度和准确性。此时,Hi-C技术是最佳选择[2]。
4)周期短、成本低、高性价比
无需构建遗传图谱,避免了繁琐的群体构建工作,在极大程度上减少了实验误差、系统偏差及机械混杂等不可控因素的干扰。无需进行大规模的基因型分型工作。相比之下,Hi-C 技术实验周期短、实验规模小,节约了时间和成本,性价比极高。
Hi-C作为安诺优达特色技术,不断探索,不断创新
不仅能揭示染色体三维结构,还可以用于辅助组装
从动物Hi-C到植物Hi-C、微生物Hi-C
从群体细胞Hi-C到单细胞Hi-C
合作单位贯穿国内外科研院所
安诺Hi-C期待您的加入……
参考文献:[1] Lajoie B R, Dekker J, Kaplan N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines[J]. Methods, 2015, 72:65-75.
[2] Burton J N, Adey A, Patwardhan R P, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions.[J]. Nature Biotechnology, 2013, 31(12):1119-25.(转化医学网360zhyx.com)
还没有人评论,赶快抢个沙发