 腾讯登录
腾讯登录自噬与神经退行性病变新进展:或许自噬也是加重病情的元凶!
| 导读 | 自噬 - 溶酶体通路维持细胞稳态并对神经性神经营养性疾病产生有益作用。最近的研究结果表明自噬可以加重该病病情,并且使细胞激活替代的高尔基体介导的降解途径,导致有毒蛋白聚集体的排出。这一过程最终导致核分解和神经元死亡。 |
聚谷氨酰胺(polyQ)障碍导致扩展的CAG三核苷酸重复进而引起的神经退行性疾病,所述重复引起蛋白质序列中的polyQ扩展。这些发现于亨廷顿舞蹈病和齿状核旁核视网膜萎缩症(DRPLA)中.DRPLA是由于atrophin1(ATN1)基因突变的结果。这种ATQ的polyQ形式会破坏对于防止聚集相关的神经退行性疾病是至关重要的,因此通过巨噬细胞自噬活化来激活核自噬过程以降解异常的核组分并恢复体内平衡。

重要的是,自噬可防止发病机制和神经退行性疾病的进展通过控制轴突稳态和消除细胞质聚集倾向蛋白。 Nucleophagy是自噬的一种特殊形式,它通过与LC3B-II的直接相互作用。然而,直到现在,核吞噬和神经退行性疾病之间的关系,尚不明确。
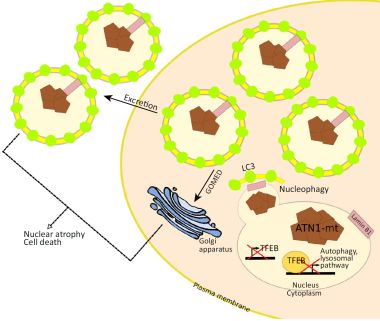
在神经退行性疾病中自噬溶酶体阻断后的替代性清除途径
最近来自Baron等人的一篇文章揭示其中的奥秘。“Current Biology”已经阐明了自噬和polyQ疾病之间的联系。作者说,典型的自噬在逐渐阻断DRLPA神经元。这导致通过细胞核的核自噬介导的和高尔基体依赖性排泄激活补偿性清除途径,其引起核变性和细胞萎缩。
在小鼠中枢神经系统神经元中表达含有65个polyQ(ATN1-65Q)的全长ATN1后,ATN1-65Q小鼠显示出强度降低和明显的有氧步态,同时随着时间的推移保持比它们的野生型对应物更活跃,突出了ATN1-65Q小鼠的逐渐运动缺陷。在3周龄时,ATN1-65Q树突状神经元中自噬体形成增加。然而,ATG5-ATG12复合体的自噬启动和自噬通量在症状性终末期ATN1-65Q小脑中受损。此外,ATN1-65Q树突状神经元累积含有未消化的底物和脂褐素的自噬囊泡。自噬溶酶体形成受损,ATN1-65Q小鼠中p62 / SQSTM1水平增加,表明自噬体清除不足,所以自噬体在末期不能与溶酶体融合.TFEB是一种促进自噬和体液两者的转录因子功能,可被ATN1 65Q转录下调。这种下调导致TFEB靶基因的诱导以与其抑制剂mTORC1减少.这些研究结果表明ATN1-65Q通过mTORC1独立抑制TFEB功能在起始和转换阶段逐步阻断自噬。
Baron等人 也观察到终末期ATN1-65Q神经元的细胞核中的病理表型。它们的细胞核的圆形形状被改变,异色区域膨胀并聚集,门富集空泡积聚在核周边。值得注意的是,用雷帕霉素和bafilomycin A1处理人类DRLPA成纤维细胞,并分别增强自噬启动或阻断自噬转换,引起核整体性改变。
鉴于这一结果,作者假设这些 对核稳定性的影响可能与所描述的自噬在核纤层蛋白B1降解中的作用有关。 根据这一想法,核纤层蛋白B1在ATN1-65Q神经元的点状细胞质区域中积累,而在野生型对应物中,核纤层蛋白B1局限于核内。 细胞质核纤层蛋白B1与polyQ聚集体和LC3B-II都共定位,这支持了核纤层蛋白B1的降解由自噬介导的理论。
Baron等人的发现有助于我们了解自噬,GOMED和神经退行性疾病中的核分裂之间的相互作用。 但是,与所有研究一样,还有几个重要问题需要解决。 首先,DRLPA中lamin B1的排泄是否存在病理学结果? 其次,ATN1-polyQ如何调控TFEB基因表达? 了解这一机制可能会导致TFEB活性的恢复,并且这可能开辟新的治疗途径来治疗DRLPA和潜在的其他神经变性疾病。
参考文献Valentin J.A.Barthet and Kevin M.Ryan.Autophagy in Neurodegeneration:Can’t Digest It,Spit It Out!(转化医学网360zhyx.com)
还没有人评论,赶快抢个沙发