 腾讯登录
腾讯登录伊沙佐米「二报二批」获CFDA批准上市,进口新药登陆中国再加速
| 导读 | 4月17日,武田宣布「枸橼酸伊沙佐米胶囊」获得国家食品药品监督管理总局(CFDA,现SDA)签发的进口药品注册证,获准联合来... |
4月17日,武田宣布「枸橼酸伊沙佐米胶囊」获得国家食品药品监督管理总局(CFDA,现SDA)签发的进口药品注册证,获准联合来那度胺和地塞米松用于治疗既往至少接受过一线治疗的多发性骨髓瘤成人患者。这是中国多发性骨髓瘤患者的首个全口服治疗方案。
根据CDE的审评概述,支持伊沙佐米在中国上市的主要依据是全球关键研究C16010,以及C16010中国延续性研究。其中,C16010中国延续性研究入组115例患者,伊沙佐米组和安慰剂组(安慰剂+来那度胺+地塞米松)的中位PFS分别为6.7和4个月,中位OS分别为25.8和15.8个月(中位随访19.8个月),ORR分别为56.1%和31%。伊沙佐米组完全缓解(CR)及非常好的部分缓解(VGPR)率为24.6%,安慰剂组为12.1%。伊沙佐米组的至进展时间长于安慰剂组(中位数分别为7.3和 4.1个月)。
伊沙佐米(Ixazomib)是一种口服、高选择性蛋白酶体抑制剂,是武田在其重磅产品硼替佐米遭遇重压之下推出的一款多发性骨髓瘤主打产品,于2015年11月20日首次获得FDA批准上市,2016/11/21获得欧盟批准。如果朋友回顾一下伊沙佐米在中国的注册上市历程,会发现这是一个非常有意思进口新药「二报二批」加速上市的案例。
如下图所示,伊沙佐米在中国的整个注册过程中,并没有出现前4位字母以“S”结尾的受理号,怎么会直接批准上市呢?
伊沙佐米中国注册审批时间轴
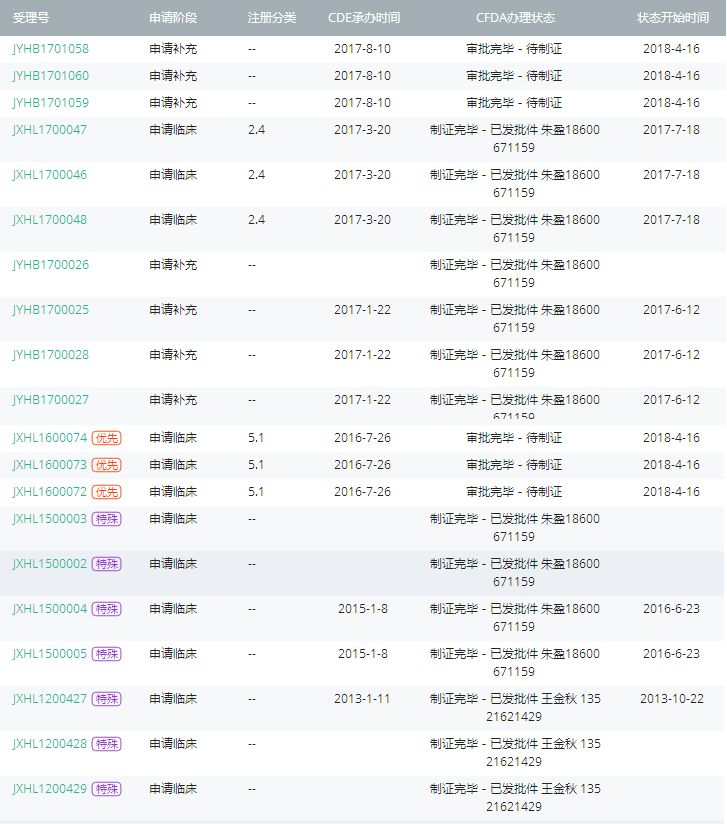
来源:医药魔方数据库--中国申报库
实际上,对进口新药而言,“三报三批”是近几年沿用的注册制度,包括:
申报国际多中心临床试验,获得国际多中心临床试验批件
申报进口药注册临床试验,获得进口药注册临床试验批件
申报进口注册(凭借“国际多中心临床试验的结果+进口药注册临床试验的批件”),获得上市资格
不过CFDA在2017年3月17日发布了《关于调整进口药品注册管理有关事项》的征求意见稿,计划将进口药的注册调整成“二报二批”,外资药企可以凭借国际多中心的临床试验数据直接申请上市(见:外资药企的春天:比二报二批更美好的事情可能要发生了)。该文件已于2017年10月10日正式实施。

其中最后一条,对于文件发布前已受理、以国际多中心临床试验数据提出免做进口临床试验的注册申请,符合《药品注册管理办法》及相关文件要求的,可以直接批准进口(不用再提交上市申请了……)。
伊沙佐米最早是于2013年1月在中国提交了国际多中心临床试验申请,走特殊审批通道,2013年10月拿到了国际多中心临床试验批件,并于2014年在国内首次登记开展了两项研究(CTR20130906,CTR20130908 ,当时的药品名称是MLN9708胶囊 )。2016年7月26日虽然再次提交进口临床申请,却包含有“以国际多中心临床试验数据申请免做进口临床试验”的内容,并且该申请(JXHL1600072/3/4)在2017/5/23被CDE纳入优先审评。
因为跳过了常规的申请上市的步骤,省掉了之前的第三报,也避免无谓的排队审评审批时间,伊沙佐米在中国的上市进程得以显著加速。2017年已经有大批进口新药凭借优先审评的激励政策加速登陆中国,随着“二报二批”的政策进一步落实,海外新药在中国上市的时间差还会进一步缩短。





还没有人评论,赶快抢个沙发