 腾讯登录
腾讯登录【研究】“谈变色变”?新冠病毒变异真实过程
| 导读 | 昨日,《国家科学评论》杂志发表文章引起热议,文章表明新冠病毒已经演化出两个亚型,其中L型的传染能力更强,各大媒体竞相报道,引起了部分群众的恐慌。事实上突变在冠状病毒这类RNA病毒中并不罕见。我们不应对文章过度解读。那么新冠病毒进化途径到底如何? |
3月2日,加州大学洛杉矶分校和中国CDC等机构合作在预印版平台bioRxiv发表的最新文章或许可以解释。

方法:研究者收集并分析了包含11株新收集于中国病人的病毒序列在内的120条新冠状病毒基因组序列。通过对这些序列进行综合分析,追踪到了多个继承性单核苷酸多态性位点(SNPs),进而确定了新冠病毒与其他冠状病毒之间的进化关系。
具体研究过程如下
研究者为了将新型冠状病毒的病理特性与特异性毒力因子联系起来,将流行病学信息与从WHO获得的序列数据进行了比较。通过比较了新型冠状病毒的感染率与近年来爆发的SARS和MERS感染率(图1A),新冠病毒的传播速度似乎比SARS和MERS快得多。为了进一步了解这一类β冠状病毒(新冠病毒与SARS同属的冠状病毒大类)的进化和追踪新型冠状病毒的突变,研究者收集了来自武汉等多个中国城市新发现患者的11株新型冠状病毒全长基因组序列并对它们进行了测序(图1C)。系统发育树表明,这11株序列与其他新型冠状病毒在进化树中能够聚集在一起,与bat-CoV-RaTG13株的同源性高于人类SARS、MERS和其他冠状病毒(之前石正丽研究员也曾报道过这一结论)。在氨基酸水平上,它们只在与人类和蝙蝠SARS中相应氨基酸序列相同的一致序列的位置上有少量随机替换。
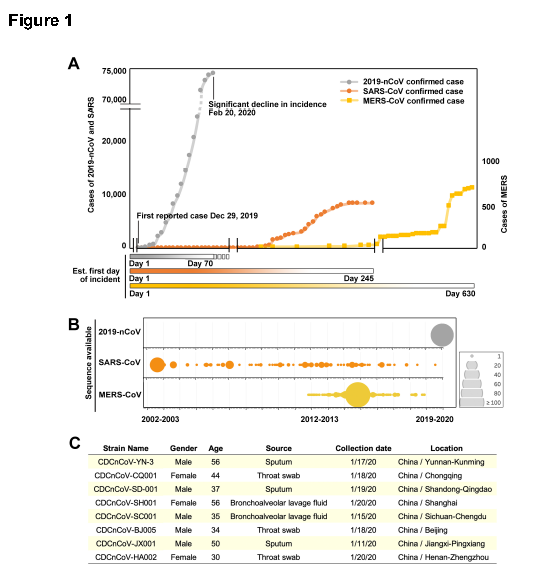
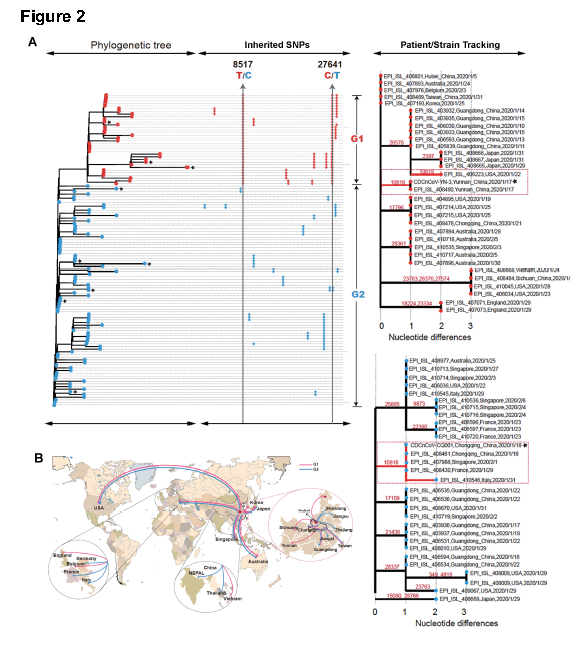
为了识别新的遗传突变,研究者以SARS-COV-2株(EPI_ISL_402125)为基础构建了包含所有120个新冠状病毒完整基因组的系统发生树。根据核苷酸读码区位点8517和27641的碱基区别,新冠病毒可分为两大类,即G1和G2。所有G1毒株在8517和27641位点分别具有胸腺嘧啶和胞嘧啶,而G2毒株的8517和27641位点的核苷酸分别为胞嘧啶和胸腺嘧啶(图2A)。流行病学资料显示,最早的G1株(EPI_ISL_406801)于2020年1月5日在武汉分离获得,而最早的G2株于2019年12月24日在武汉分离获得。这说明该两类亚型在同一城市形成了共流行的局面。而在每一类型中,研究者还观察到多个毒株中附加的共同突变。研究者基于这些可遗传突变和毒株分离的时间及位置生成了一个突变树图,这有助于我们追踪病毒的传代关系和流调分析。比如说1月10日至15日在广东鉴定的5株毒株在G1背景下的28578核苷酸位置均具有相同的突变,表明其可能由同一人传播。具有类似突变的毒株也分离于1月29日至31日在日本发现的3例患者,不同的是,这些毒株在核苷酸位置2397处具有另外附加突变,而于1月22日分离自美国的毒株则在核苷酸位置10818处具有附加突变。而该位点在G1和G2毒株中发现了共享突变,这值得我们关注。而目前的分析也表明G1和G2毒株已传播到大多数国家和地区。
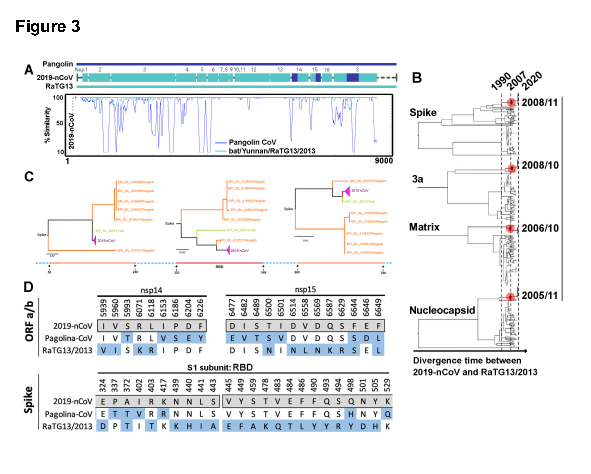
与新型冠状病毒最密切相关的β冠状病毒是RaTG13,该病毒于2013年分离自云南的蝙蝠中(图3A)。研究者用核苷酸序列对特定病毒蛋白如orf1a、spike、matrix和核衣壳进行了额外的系统发育分析(图3B),发现RaTG13株与其他蝙蝠SARS样冠状病毒株有同样的密切关系。由此研究者进一步估计,新型冠状病毒和RaTG13之间的大多数蛋白质差异发生在2005年和2012年,而人类SARS和蝙蝠SARS样冠状病毒之间的差异发生在1990-2002年(图3B)。比较全长spike蛋白序列时,新型冠状病毒与人和蝙蝠SARS的序列同源性为39%,与MERS或其他冠状病毒的序列同源性为29%。值得注意的是,我们发现新型冠状病毒和穿山甲冠状病毒在棘突蛋白的RBD(aa 315-550区域)中共享几乎相同的氨基酸序列,但在RaTG13中不共享(图3A和3D)。虽然2019-nCoV在整个基因组结构中与RaTG13最同源,但spike蛋白的RBD与穿山甲冠状病毒最同源,这表明在2019-nCoV的进化过程中,RaTG13样株和穿山甲冠状病毒样株之间可能发生重组。研究者检查了基因组中所有的氨基酸突变。结果发现,当比较穿山甲冠状病毒和新型冠状病毒时,除了RBD,非结构蛋白14和非结构蛋白15中的区域也共享连续序列(图3D)。
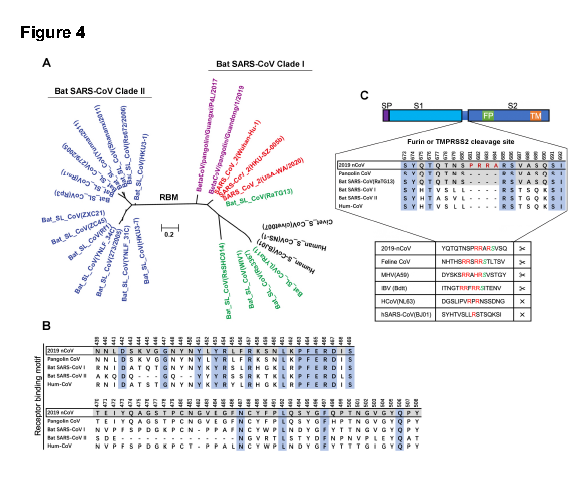
同时该研究还表明,新冠病毒在spike蛋白内核苷酸23619-23632位特异性插入4个氨基酸(681-PRRA-684)(图4C)。该插入氨基酸在新型冠状病毒的spike蛋白中为哺乳动物furin蛋白创建了一个潜在的裂解位点RRAR。潜在的furin裂解位点插入到spike蛋白的S1和S2结构域之间的边界,PRRA插入的第一个脯氨酸残基可能将β转化引入到多肽链中。那么这种插入是否只存在于新冠病毒中呢?研究者使用来自人类、果子狸和蝙蝠的SARS冠状病毒代表株进行了序列比较。结果表明,这种插入是新型冠状病毒独有的(图4C)。已有研究表明,在SARS冠状病毒中引入一个furin裂解位点,将导致了荆棘蛋白的裂解,从而会增强该病毒的膜融合活性。此外,在SARS冠状病毒模型中引入一个被切割的荆棘蛋白,它可以直接进入宿主细胞。而新型冠状病毒spike蛋白被认为是与ACE2受体相互作用的关键部分。因此,改变spike蛋白S1-S2亚单位的突变或插入删除会显著影响病毒感染。PRRA的插入已经证实就是位于S1S2亚基的交接处,可以猜想这能使spike蛋白进入furin过程,从而触发病毒融合事件。
研究者鉴定了新型冠状病毒不同毒株间所共同拥有的多个单核苷酸多态性位点(SNP)。发现新型冠状病毒基因分为两个不同的组,在8517和27641位点具有不同的核苷酸多态性。尽管这目前尚不知这两组不同的新型冠状病毒是动物传给人类之前或之后进化形成的,但这两组新型冠状病毒都能够在疫情早期在武汉的样本中检测到,之后不同的基因型的新型冠状病毒又在其他地区被分别同时检测到。通过结合新型冠状病毒后代中额外的可遗传突变与患者样本收集的时间和地点,可以追踪的病毒传播途径。
研究者发现除了棘突蛋白的受体结合区更接近于广东17年分离的穿山甲冠状病毒外,新型冠状病毒与RaTG13在整个基因组中具有高度的同源性。基于这些发现,我们假设在新型冠状病毒的进化过程中,RaTG13和穿山甲冠状病毒株之间可能发生棘突蛋白RBD重组。
研究者预测Ⅰ类冠状病毒(包括新型冠状病毒)可以通过ACE2感染宿主细胞,而分支Ⅱ冠状不能通过ACE2感染宿主细胞。根据现有的基因组序列,分支Ⅱ蝙蝠冠状病毒(超过49个染色点)比分支Ⅰ蝙蝠冠状病毒(大约12个毒株)多。同源β冠状病毒是否能通过RBM组来改变嗜性将是一个值得探讨的问题。同时还发现了一个独特的由4个氨基酸(PRRA)组成的在棘突蛋白的S1和S2结构域之间的插入,这可能是一个furin裂解位点。因此,研究者假设PRRA的插入可能触发病毒融合事件。这将为迅速制定新的策略来治疗或预防提供参照。
参考文献:https://www.biorxiv.org/content/10.1101/2020.02.29.971101v1
(转化医学网360zhyx.com)



还没有人评论,赶快抢个沙发