减数分裂是有性生殖生物生命周期中的一个重要阶段,二倍体生殖细胞经由这一过程转化为单倍体的配子,构成了遗传多样性的基础。其核心机制是同源染色体重组,在此过程中,程序性DNA双链断裂(DNA double-strand breaks,DSBs)被修复,从而形成染色体精准分离所必需的交换(crossover)。重组异常往往导致配子发生失败或产生非整倍体配子,造成生育能力低下、不育、流产或出生缺陷,甚至肿瘤的发生。
非梗阻性无精子症(non-obstructive azoospermia,NOA)和早发性卵巢功能不全(premature ovarian insufficiency,POI)是不育症和生育力低下的临床常见原因,二者均为高度异质性疾病,而异常的减数分裂重组在其中发挥着重要作用。虽然目前已发现众多与人类NOA和POI患者减数分裂重组异常相关的基因突变,但对配子发生失败的遗传机制仍然知之甚少。
近日,中信湘雅生殖与遗传专科医院卢光琇、林戈教授团队谭跃球课题组在Human Reproduction Update杂志上发表了题为“Meiotic recombination: insights into its mechanisms and its role in human reproduction with a special focus on non-obstructive azoospermia”的综述性文章。该文通过梳理近几十年来国内外相关研究进展,结合课题组研究成果,全面系统总结了减数分裂同源重组的分子机制,深入剖析了重组缺陷与配子发生失败间的关系,整理了迄今为止已发现导致NOA和POI的基因,并基于此对疾病的个性化诊疗方案进行了阐述。
要点速览
减数分裂过程中的结构组件
染色体轴和联会复合体的拉链状中央成分(central element,CE)是减数分裂同源染色体重组过程中的2种至关重要的高度保守的蛋白质支架结构。前者为染色体复制提供支架,并控制DSB的形成和同源染色体间交换的形成,包括黏结蛋白复合物、HORMA结构域蛋白(HORMAD1和HORMAD2)和轴元件(AE)蛋白质SYCP2和SYCP3;后者主要介导同源染色体轴之间的锚定关系,并提供结构框架以促进重组位点的交换。
减数分裂的早期事件:DNA双链断裂
减数分裂重组起始于程序性DSB。DSB位点并非沿染色体随机分布,而是在局部染色质环上由PRDM9介导的H3K4me3和H3K36me3确定的热点区域,从而保证交叉重组出现在恰当的位置,避免富含重复序列的基因组区域内发生重组以及由此引起的非等位基因同源重组或基因组重排。染色质上PRDM9结合的DSB热点与未联会染色体轴之间进行联结,并在SPO11-TOPOVIBL复合物及相关辅助因子作用下,进一步诱导形成DSB。此外,DSB发生的频率也受到严格调控,以最大程度上避免负载过大可能导致的危害,此过程主要通过DSB激活损伤反应激酶ATM,通过负反馈途径抑制SPO11蛋白的活性来进行调控。
重组中间体的形成
DSB形成后,需经MRN复合物协同核酸内切酶CtIP、解旋酶BLM以及核酸外切酶DNA2和EXO1进行切割加工,产生具3’末端的游离DNA单链;后者与以RPA复合物为主的蛋白结合,以阻止单链DNA的降解或形成二级结构。随后,RAD51/DMC1取代RPA,与ssDNA结合,引发ssDNA入侵dsDNA,并形成D-loop。D-loop形成后,首先第一个入侵的DSB末端引发DNA合成,使D-loop得以延长,随后延长的D-loop在MEIOB–SPATA22的介导下捕获第2个DSB末端,形成经典的dHJ中间体。
基因交换:交换和非交换
dHJ中间体可以通过dissolution或resolution两种方式进行解开,如果dHJ通过dissolution方式解开则会产生非交换产物,如果以resolution方式解开则可形成交换或非交换产物。交换产物的形成使得同源染色体在双线期形成交叉,但是DSB大多数是以非交换形式进行修复的。交换有两类,即由MutLγ蛋白切割dHJ结构而产生的第一类交换和由结构选择性内切酶Mus81-Eme1、SLX1-SLX4/BTBD12和GEN1切割重组中间体所产生的第二类交换;第一类交换是减数分裂同源染色体发生遗传物质重组的主要方式。
减数分裂检验点网络及其在配子发生中的作用
减数分裂进程受到细胞周期检验点监控,主要有三个不同的减数分裂检验点,即DNA损伤检验点、联会检验点和纺锤体检验点,负责监测异常的染色体联会、配对和重组,并触发细胞周期停滞。前两种也被称为粗线期检验点,保证生殖细胞染色体联会和重组的正常进行。纺锤体检验点确保同源染色体的准确分离。
除减数分裂基因缺陷外,染色体异常也可能导致异常配对或联会,并在粗线期生殖细胞中产生不同的染色体构象,包括单价体、异形二价体和多价体。
减数分裂异常引发的NOA及其临床治疗
NOA主要由减数分裂基因突变引起,文章总结了26个基因导致人类NOA的基因。鉴定与减数分裂重组相关的致病基因不仅有助于临床医生的诊断,而且为不孕症患者的最终治疗提供了一个精确的靶点。
文章精读
(*以下内容阅读大约需要10min)
目录
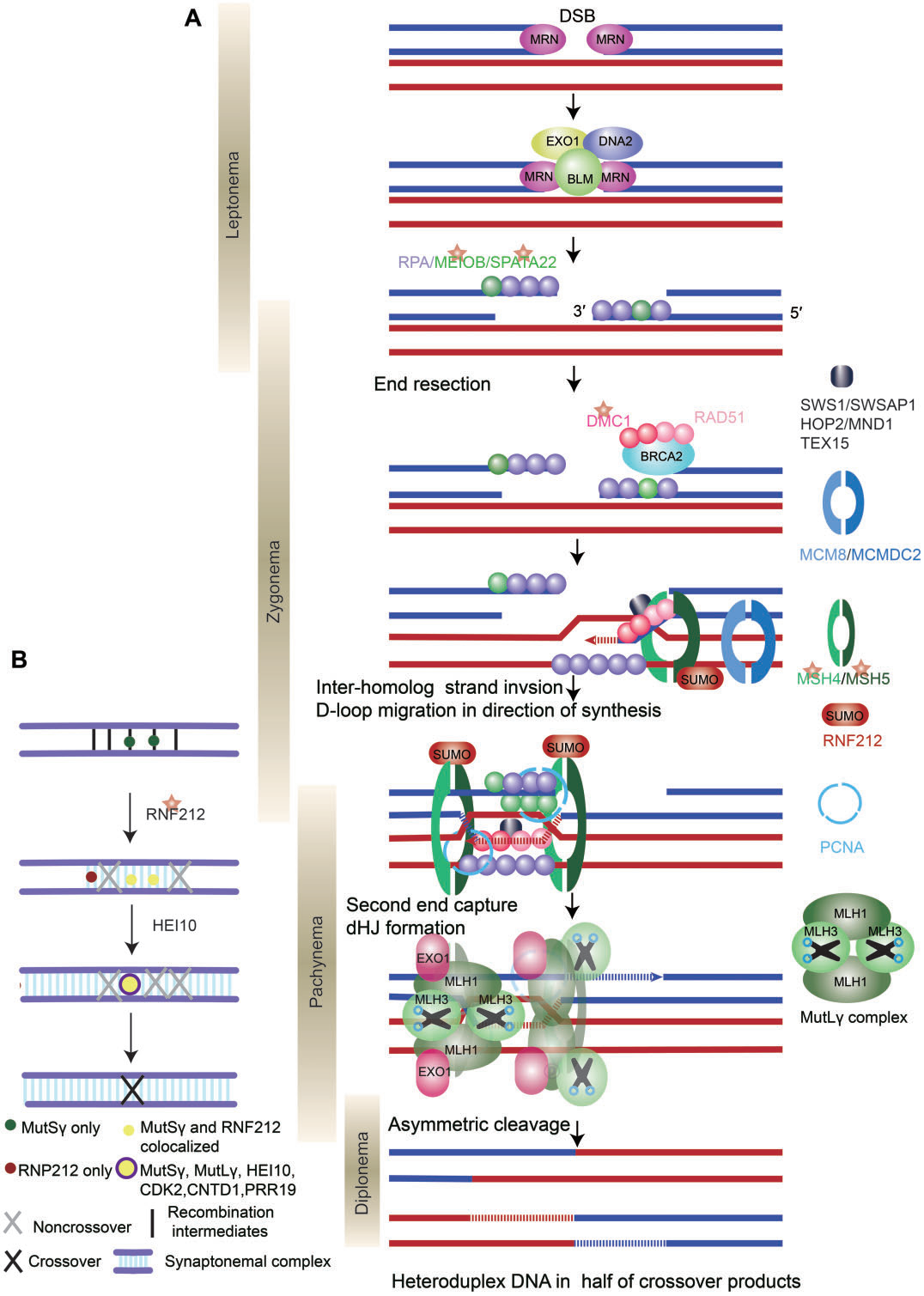
减数分裂起始于独立的减数分裂前S期,随后为延长的第一次减数分裂前期(前期I),该阶段根据染色体的行为,可进一步分为细线期(leptotene)、偶线期(zygotene)、粗线期(pachytene)、双线期(diplotene)和终变期(diakinesis)(图1A)。在前期I,由细线期阶段保守的SPO11-TOPOVIBL复合物介导的程序性DSB启动同源染色体重组。在粗线期,这些DSB被修复以产生交换或非交换,其中交换是保证同源染色体在减数分裂I中精准分离的必要条件。在减数分裂前期I的任何阶段发生的错误都会导致异常的同源染色体配对、联会或重组,引发减数分裂停滞或染色体分离缺陷。
图1. 减数分裂染色体行为和减数分裂DNA双链断裂(DSBs)示意图
减数分裂过程中,不同结构组件的联结和解离驱动着染色体和染色质构象的动态变化。其中,有两种高度保守的蛋白质支架结构对减数分裂重组过程尤为重要,一个是染色体轴,包含黏结蛋白复合物、HORMA结构域蛋白和轴元件(axis element,AE)蛋白质SYCP2和SYCP3(图1),为将复制染色体排列成环的线性阵列以及控制DSB形成和同源染色体间交换形成提供支架。另一个是联会复合体(synaptonemal complex,SC)的拉链样中央成分(central element,CE),介导同源染色体轴之间的锚定关系,并提供结构框架以促进重组位点的交叉。
黏结蛋白复合物
黏结蛋白复合物是一种包含四个核心亚基的环状结构:SMC1蛋白家族的2个亚基SMC1 α/β和SMC3,一个较小的非SMC蛋白亚基SA1/SA2或STAG3,以及一个kleisin亚基REC8、RAD2I或RAD21L(图1B和C)。其中,SMC1β、STAG3、REC8和RAD21L在减数分裂中特异性表达。该复合物在染色体轴形成、环-轴组织、姐妹染色单体连接、端粒完整性、同源染色体配对/联会、同源染色体重组和分离中发挥着重要作用。
HORMAD1和HORMAD2
HORMA结构域蛋白,包含HORMAD1和HORMAD2,在减数分裂前期I优先沿未联会染色体轴共定位,对同源染色体相互作用的调节和同源染色体联会的监测至关重要。HORMAD1被减数分裂黏结蛋白复合物REC8和RAD21L以及AE蛋白SYCP2和SYCP3募集。而HORMAD2则可能被HORMAD1和/或SYCP2招募到轴上。联会后,HORMAD1/2通过TRIP13介导的HORMAD N末端结构域暴露机制从联会染色体上移除。TRIP13识别N端结构域的过程尚不清楚。
同源染色体的配对和联会复合体的形成
重组涉及同源染色体之间的配对或识别。在从细线期到偶线期的进程中,端粒附着在核膜上,形成一个类似花束的形状(称为花束期),并沿核膜移动,以缩短染色体之间的相对距离,提高同源染色体之间的相互搜索效率,促进轴元件列对和染色体配对。三种蛋白质复合物参与其中(图1A):LINC复合物包括SUN1和KASH5,shelterin复合物和TERB1-TERB2-MAJIN复合物。
染色体配对促进了列对同源染色体之间的SC组装。SC是不同物种间高度保守的蛋白结构,主要由AEs或侧成分(lateral elements,LEs)、横向轴丝(transverse filaments,TFs)和中央成分(Central element,CE)三部分组成。SYCP2和SYCP3是AE/LE的两种主要组分,二者相互作用形成染色体轴核心,为染色质环的形成提供了一个柔性支架,并促进染色体轴的组装。TFs的主要组分是SYCP1;CE蛋白可分为三组,包括起始蛋白SYCE1、SYCE3和SIX6OS1,延伸蛋白SYCE2和TEX12,以及稳定蛋白SCRE。
SC组分触发沿配对同源染色体全长的拉链状自组装,这时的SC可以被看作是一个通过蛋白质-蛋白质聚合堆叠的刚性结构(图1A和1D)。最新研究表明,SC具有类似液体的特性,可在空间上调节减数分裂重组因子。这一特征也可能为减数分裂染色体和染色质动力学提供基础。
02 减数分裂的早期事件:DNA双链断裂
减数分裂重组起始于程序性DSB。DSB位点并非随机分布,而是定位于由组蛋白3甲基化标记(H3K4me3和H3K36me3)的染色质环上的热点区域,这一过程经由PRDM9催化。
DSB热点位置确定
在真核细胞中,大部分DNA被包裹缠绕在组蛋白八聚体周围形成核小体,核小体是染色质的基本结构单元,也是DSB形成的物理屏障。DSB热点由DNA结合蛋白PRDM9和染色质重塑因子HELLS定位。PRDM9和HELLS形成了一种先驱复合物,可构建开放的染色质,增加了其他核小体重塑因子、组蛋白修饰因子和DSB形成机器的可及性(图2)。
图2. 减数分裂DNA双链断裂(DSB)蛋白的作用模型
热点位点与未联会染色体轴连接
染色质环上PRDM9结合的DSB热点与未联会染色体轴之间的相互作用是DSB形成所必需的(图1D),至少包含三种机制:(i)EWSR1与PRDM9和磷酸化REC8相互作用介导DSB位点与未联会染色体轴之间的连接(图2);(ii)PRDM9直接与染色体轴上的黏结蛋白相互作用;(iii)CXXC1通过与PRDM9和轴相关蛋白IHO1相互作用,作为一个适配器将DSB位点锚定在染色体轴上。这些过程中的任何一个缺陷都仅导致DSB形成的部分失败,表明功能上的冗余。
SPO11-TOPOVIBL和辅助因子催化DSB形成
DSB的形成是由位于染色体轴上的SPO11-TOPOVIBL复合物催化的,主要有两种SPO11亚型:SPO11α和Spo11β,前者2号外显子缺失,后者为全酶。SPO11β参与从细线期到偶线中期常染色体DSB的形成,而SPO11α则负责在偶线中期之后的假常染色体区域(pseudoautosomal region,PAR)DSB的产生。部分进化保守的减数分裂特异性pre-DSB重组体REC114、MEI1、MEI4、ANKRD31和IHO1,可能负责将SPO11-TOPOVIBL靶向到PRDMP依赖的DSB热点(图1D)。这些重组体与未联会染色体轴的锚定依赖于HORMAD1-IHO1相互作用,但其协调SPO11-TOPOVIBL加载或激活的机制尚不清楚。在PAR中,DSB的形成很大程度上独立于PRDM9,pre-DSB重组体的积累独立于HORMAD1,相反依赖于特定类型的重复DNA序列。
DSB数量和分布的调控
DSB的数量必须严格控制,以避免DSB负荷过大而导致联会和DSB修复中的严重缺陷。ATM激酶由DSB激活,通过负反馈途径确保每个热点上进行最小的SPO11切割,一旦该酶缺失,DSB大量产生,造成精母细胞在前期I停滞。近期还发现可通过破坏IHO1来负调节DSB,防止DSB过度产生。
DSB很少出现在着丝粒周围和异染色质内。如果在着丝粒周围形成大量DSB,它们可能以交换的形式被修复,从而破坏动粒定位或着丝粒连接,干扰染色体分离,并导致出生缺陷,如21三体。动粒蛋白复合体可能通过改变局部染色质组织来干扰SPO11激活因子的可及性,从而抑制着丝粒周围有害DSB的发生。此外,着丝粒周围异染色质蛋白HP1和黏结蛋白复合物在防止DSB激活因子的募集中起着关键作用。富含重复序列的异染色质区域内的DSB是减数分裂非等位基因同源重组诱导的基因组不稳定性的潜在来源,可能是导致染色体易位的潜在机制。
03 重组中间体的形成
DSB末端经酶切,产生具有3’末端的游离DNA单链(ssDNA)。ssDNA末端与重组酶结合生成前导纤维(presynaptic filament),然后进行同源DNA序列的搜索和链交换,以物理连接一对同源染色体的两个非姐妹染色单体。链交换涉及单链和多链交换,触发同源染色体之间的联合分子(joint molecules,JMs)或重组中间体的形成(图3)。形成典型重组中间体的单链交换事件包括三臂和排列,例如D-loop(ssDNA置换一条同源dsDNA)和双Holliday junctions(dHJs)。多链交换形成复杂的多条染色体重组中间体,包括三个或四个相互连接的DNA,如三元JM和四元JM。

图3. 交换/非交换形成机制
DSB形成后,进化上保守的MRE11-RAD50-NBS1(MRN)复合物,协同核酸内切酶CtIP、解旋酶BLM和核酸外切酶DNA2和EXO1,切割DSB末端形成3’突出末端(图4A)。然后,ssDNA末端与RPA、MEIOB和SPATA22等蛋白质结合,以阻止ssDNA的降解或形成二级结构。
图4. DNA双链断裂交叉和修复的分子机制
随后,重组酶RAD51/DMC1取代ssDNA结合蛋白,通过单链侵入或单端侵入过程促进ssDNA入侵同源染色体。促进重组酶RAD51和DMC1在RPA包被的单链DNA上组装的因子称为重组介体蛋白,包括BRCA2、MEILB2/HSF2BP、BRME1/MEIOK21、SWS1和SWSAP1。
单端侵入产生异源双链DNA,形成D-loop。DNA聚合酶以入侵单链的3’端作为引物催化DNA合成,使D-loop得以延长并被RPA结合。这代表了RPA除了结合3’悬末端外的另一个作用。由于D-loop的基本元素是同源中间体,因此需要各种机制来确保减数分裂重组过程中的同源性偏倚;重组酶DMC1是同源染色体搜索中的一个关键因素,而在有丝分裂中介导同源姐妹染色单体间重组的RAD51的链侵入活性在减数分裂中受到抑制。
分支迁移、第二末端捕获和dHJ的形成
D-loop形成后,发生分支迁移(D-loop沿DNA移动),维持D-loop稳定。在小鼠中,D-loop迁移导致只有一半的交换产物在起始DSB的一侧形成异源双链DNA(图4A)。
大多数D-loop(在小鼠中为90%)通过合成依赖性链退火(SDSA)机制解离,并发展为早期非交换(NCO)。一小部分D-loop形成稳定的中间体,向DNA合成方向迁移。重组中间体的稳定需要一系列蛋白质,包括SHOC1、TEX11、SPO16和MCMDC2(如表III所示),进而引导重组中间体发生交换。最终,MEIOB-SPATA22和RPA之间的相互作用以剂量依赖的方式促进第二末端捕获和dHJ的形成(图4A)。
04 基因交换:交换和非交换
重组中间体可以通过dissolution或resolution两种方式进行解离。resolution可以形成交换和/或NCO,而dissolution仅导致NCO(图3)。在小鼠中,最初产生了200-300个DSB,但只有10%出现交换,其余均为NCO。
交换
双线期阶段末期,同源染色体通过交叉进行物理连接。交换对于确保遗传多样性、保持基因组稳定性和促进适应性进化至关重要。交换模式(存在、数量和定位)受到严格控制,并表现出性别二体性。染色体轴长度在调节交换模式中起着重要作用。女性的染色体轴比男性长,因此能产生更多的DSB和交换事件。DSB热点的使用在男性和女性中也有所不同,男性交换更多集中于亚端粒区域。正常交换遵循三个原则:强制交换确保每对同源染色体至少形成一个交换;交叉干扰确保交换事件在染色体上均匀分布;交叉稳态确保交换数的稳定性。在人类中,男性的交换干扰更强,这在一定程度上解释了交换分布中的性别二体性。
交换可以分为两类,I类交换和II类交换
I类交换占所有交换事件的90-95%,并且只依赖于ZMM蛋白,包括HFM1、MSH4、MSH5、RNF212和HEI10。ZMM蛋白的缺失导致交换形成不足。ZMM蛋白保护dHJs免受BTR(BLM-TOPIIIa-RMI1/2)“解离酶”复合物介导的解离,并通过一个称为“交换指定”过程协调DSB向交换修复(图4b)。PRR19-CNTD1复合物也参与这一过程,促进减数分裂交换分离和成熟。最后,MutLγ在MutSγ、RFC、PCNA和EXO1的帮助下,不对称切割dHJ,以实现仅产生交换的解开。MutLγ的作用机制尚未明确阐明。
与I类交换事件不同,II类交换事件是随机发生的,二者在不同物种间所占比例不同。在高等哺乳动物减数分裂期间,II类交换通常受到抑制,但在其他一些生物中,如酿酒酵母,几乎所有同源染色体交换都涉及II类交换。II类交换依赖于结构选择性内切酶,包括MUS81-EME1、SLX1-SLX4/BTBD12和GEN1,产生交换产物和NCO产物。结构选择性内切酶可以提供一种保障机制,确保未被MutLγ解开的JMs在染色体分离之前被处理。
非交换
减数分裂过程中产生的DSB数量远远超过交换事件。未形成交换的重组事件产生NCO,NCO在同源染色体配对和交叉稳态中起着重要作用,NCO可通过三种机制形成(图3)。大多数是通过SDSA途径分解D-loop形成的。或者,未受ZMM保护的dHJs可被BTR复合物解开,只形成NCOs。未受ZMM保护且未被BTR解开的dHJs,以及复杂的JM结构,可以通过结构选择性内切酶分解为NCOs。
综上所述,重组中间体resolution或dissolution产生的NCOs,以及dHJ分离产生的I类交换,确保了同源染色体准确配对和分离。
05 减数分裂检验点网络及其在配子发生中的作用
配对、联会和重组缺陷通常与配子生成失败或非整倍体生殖细胞有关,导致生育能力低下/不育或出生缺陷。三个不同的减数分裂检验点,即DNA损伤检验点、联会检验点和纺锤体组装检验点,负责监测这些错误并触发细胞周期停滞。前两种也被称为粗线期检验点,保证了生殖细胞联会和重组的正常进行。纺锤体组装检验点确保同源染色体的准确分离。
DNA损伤检验点
DNA损伤检验点是一种重组依赖的阻滞机制,当DSBs在粗线期不能完全被修复时启动细胞凋亡。DSB检测依赖于两种高度保守的蛋白激酶,ATM和ATR。原则上,未修复的DSB激活ATM和ATR,进而分别激活效应激酶CHK2和CHK1,并可通过p53/p63依赖性凋亡途径促进减数分裂停滞。
联会检验点
联会检验点沉默未联会染色质上的基因,并清除携带异常联会的减数分裂细胞(联会错误)。在偶线期向粗线期的转变过程中,未联会常染色体轴上的SYCP3和HORMAD1/2促进BRCA1、ATRIP、TOPBP1和ATR的募集,而募集的ATR促进H2AX(γH2AX)的磷酸化。然后,MDC1,一个与TOPBP1相互作用的γH2AX阅读器,促进ATR、TOPBP1和γH2AX在整个非联会染色质区域扩展,最终形成一个较大的γH2AX区,并导致非联会染色质的减数分裂沉默(MSUC)。当MSUC被激活时,常染色体中减数分裂进程所必需的基因被沉默,生殖细胞的凋亡被触发。
减数分裂性染色体失活(MSCI)是一种仅限于男性的MSUC拓展,参与XY小体内性染色体非联会区域的转录抑制。X和Y染色体仅在PAR中发生联会,而未联会性染色体区域包含“粗线期致死”基因,如ZFY。这些基因的表达使减数分裂进程停滞在粗线期。大部分未联会的X染色体上的γH2AX标记将TRIM28募集到XY小体,与组蛋白甲基转移酶SETDB1相互作用,诱导H3K9me3促进异染色质形成和基因沉默(图5)。MSCI还可以诱导ATR介导的DNA损伤应答因子(如BRCA1、ATR和MDC1)从常染色体分离到未联会性染色体,以防止常染色体上过度的DNA损伤反应活性,并确保减数分裂的正常进程。因此,有缺陷的MSCI与粗线中期的完全减数分裂停滞和精母细胞清除有关。
染色体异常引起的异常联会
除减数分裂基因缺陷外,染色体异常还可能导致异常配对或联会,并在粗线期生殖细胞中产生不同的染色体构象,包括单价体、异形二价体和多价体。
单价染色体,如XO粗线期卵母细胞,可以通过自我联会形成完全联会的发夹结构,以避免MSUC引发的沉默。不自我联会导致MSUC激活,沉默X染色体上的基因并诱导生殖细胞凋亡。
由重排(如插入)引起的衍生染色体可以与同源染色体形成异形二价体。一些不能与同源染色体联会的染色质形成非联会染色质环,进而激活MSUC。在粗线期精母细胞中,这些染色质通常并入XY小体中,导致环上的基因沉默。非同源染色体联会或联会调节允许染色质绕过MSUC,但会干扰染色体分离或激活纺锤体组装检验点。
在多价构象的情况下,完全同源或非同源联会可以绕过MSUC。然而,染色体重排断点周围多价结构的空间限制可能会阻止减数分裂重组蛋白的进入,导致联会和重组失败。在精母细胞中,多价构象会干扰XY小体,导致不育。
06 减数分裂异常引发的NOA及其临床治疗
全球大约10-15%的育龄夫妇受不孕症困扰,大约50%是由男性因素导致。其中,无精子症,包括梗阻性和非梗阻性,被认为是临床上最严重的男性不育表型。NOA可累及0.6%普通人群男性和10%的不育男性。
NOA主要由减数分裂基因突变引起。目前已确定大量基因可以作为男性生育能力低下或不育的预测标志物。据报道,60多个精母细胞基因已被证明会影响小鼠的生育能力,其中至少有26个基因会导致人类NOA(见下表)。
鉴定与减数分裂重组相关的致病基因不仅有助于临床医生的诊断,而且为不孕症患者的最终治疗提供了一个精确的靶点。然而,由于配子发生损伤的不可逆性,减数分裂相关不孕不育患者的生殖机会极其有限。
目前,NOA患者只能通过TESE结合后续ICSI技术获得自己的生物学后代。现有文献表明,几乎所有由减数分裂重组相关基因突变引起的NOA患者都表现出精母细胞停滞,不符合TESE-ICSI的条件,且成功性很低。因此,减数分裂重组相关基因突变鉴定可作为一种无创性诊断分子标记,用于指导NOA患者TESE-ICSI的必要性。
基因组编辑是帮助减数分裂相关NOA患者获得正常配子的一种很有前景的技术。减数分裂停滞不会影响精原干细胞(SSC)的有丝分裂过程,移植的SSC可以通过血-睾丸屏障转移到基底室,完成正常的精子发生,因此,SSC移植可以治疗由减数分裂突变引起的男性不育。目前应用最广泛的基因组编辑技术是CRISPR/Cas9。
然而,为了实施生殖系基因组编辑,有必要解决有效性和安全性问题,以及潜在的伦理和法律限制。例如基因编辑技术的非靶向效应;SSC在体外培养环境中可能发生的表观遗传修饰变化和新的基因突变;而高额成本也大大降低了其可行性。此外,基因编辑技术还可能会导致NOA患者遭受耻辱和歧视。因此,目前没有法律批准人类生殖系基因组编辑。
07 减数分裂重组缺陷引发的POI
减数分裂重组在精子发生和卵子发生中的过程本质上是相同的,许多导致NOA的减数分裂相关基因也会造成POI(见下表)。因此,NOA患者一旦鉴定出了此类基因缺陷,则有必要对女性家庭成员进行相关基因筛查。此外,减数分裂重组中的变异与卵泡池的早期或后期消耗有关。当在女性中检测到NOA相关基因突变时,临床医生应与之讨论POI或卵巢功能障碍的风险,并尽早帮助其做出有关生育能力保存或妊娠的决定。
严重POI患者最常见的治疗方法是卵子捐献。此外,新的POI治疗方法也正在探索中。特别是在不改变基因组的情况下,通过引入外源性互补RNA(cRNA)来增加原始卵泡数量,可以改善卵母细胞的遗传缺陷。另一个潜在的治疗策略是增强卵泡活性和延缓生殖衰老。其他技术,如干细胞治疗和抑制卵巢血管生成,已被研究以帮助卵泡池衰竭或POI患者恢复生育能力。
08 减数分裂异常而合并遗传性癌症
值得注意的是,一些减数分裂相关基因,尤其是DNA修复相关基因,不仅是NOA和POI的常见原因,也与遗传性癌症相关。如,NOA致病基因MLH1和ATM,以及POI致病基因BRCA2和MCM9都已被证实可以增加癌症遗传易感性。因此,在NOA和POI患者中识别出此类减数分裂相关缺陷,应建议进行常规癌症监测。
小 结
该综述通过全面梳理减数分裂重组新进展,归纳阐述了减数分裂重组和人类NOA的分子机制。减数分裂重组过程有很多蛋白和基因参与其中并相互作用,构成了复杂的分子网络。而这篇综述提供的信息将有助于对这些基因的功能进行解析,随着体外配子发生技术的发展和基因重组编辑技术的安全性和有效性的提高,对此类疾病的安全有效治疗或将成为可能。
谢春波研究生为该论文的第一作者,中南大学谭跃球教授为通讯作者,中南大学林戈教授和卢光琇教授以及浙江大学陆林宇教授为论文提供了指导,为重要并列作者。
 腾讯登录
腾讯登录












还没有人评论,赶快抢个沙发