 腾讯登录
腾讯登录简化甲基化测序 (RRMS) 可捕获 100% CpG 岛且不止于此
| 导读 | 该方法可靶向基因组中的重要区域,从而能以经济高效的方式,对感兴趣样本表征全基因组的甲基化模式。 |
联系方式:publications@nanoporetech.com
更多信息请访问:www.nanoporetech.com 和 publications.nanoporetech.com

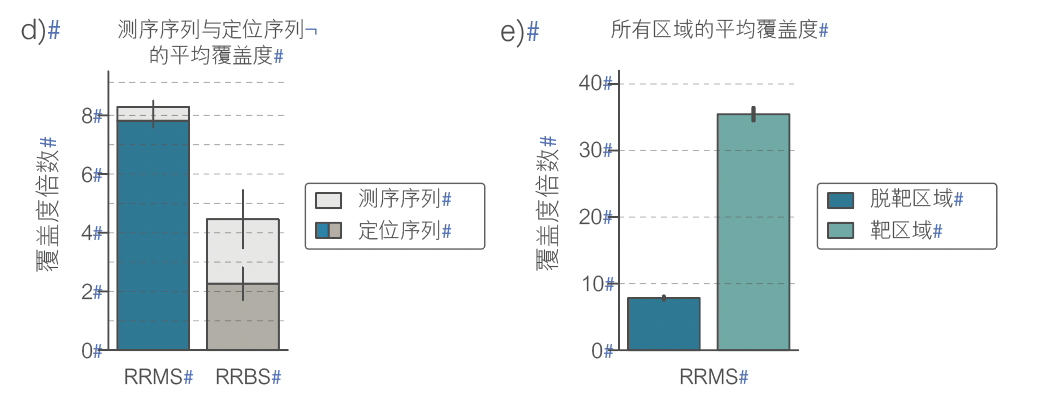


图 1 采用 AS 的 RRMS a)读长长度分布 b)Bed 工作流程 c)特征序列 d)测序序列对比定位序列 e)覆盖度 f)示例区域 g)与 RRBS 重叠 h)已识别的 CpG i)与 RRBS 的相关性
自适应采样可增加靶向序列的覆盖度,因此仅用一张 MinION™测序芯片就能识别基因组关键区域中8M的高置信度CpG,并且准确度高
纳米孔测序能够直接检测甲基化胞嘧啶(例如在 CpG 位点),而无需亚硫酸氢盐转化。CpG 位点常出现在称为 CpG 岛 (CGI) 的高密度簇中。>60% 的人类基因的启动子是嵌入在 CGI 区域的。启动子内甲基化模式的变化与基因表达的变化相关。简并代表性亚硫酸氢盐测序 (RRBS) 无需测序整个基因组,即可实现全基因组甲基化分析,但此方法成本高昂且耗时。此外,其文库制备方法复杂,精确度不高,不能靶向任何特定启动子区域。自适应采样 (AS) 是一种快速、灵活、精确的方法,可在测序过程中去除脱靶区域,从而富集感兴趣区域(例如 CGI)(图 1a),无需预先处理样本。此处,我们将 Oxford Nanopore的甲基化检测方法与 AS 相结合,并在两对肿瘤/正常细胞系中将该方法的性能与 RRBS 进行比较。我们使用图中所示工作流程(图 1b)制备 AS 靶向区域文件,覆盖 310 Mb 的基因组,包括约 28000 个CpG 岛和其他关键特征序列(图 1c)。AS 保留的数据比例高于 RRBS,且在靶区域的读长序列比例更高(图 1d 和 e)。AS 显示出比 RRBS 更均匀的覆盖度(图 1f),恢复了更多的 CpG(图 1g 和 1h),并且可重现性高(图 1i)。




图2 使用 RRMS 检测的肿瘤/正常细胞差异 a)工作流程 b)DMR c)CpG 岛 d)和 e)5mC 和 5hmC 的分布,f)示例区域 g)靶区域的读长序列 h)CNV i)拷贝数/染色体
可以探索不同样本之间的甲基化模式,以识别大量差异性甲基化的特征序列。丢弃的读长序列也可用于识别拷贝数变异 (CNV)







还没有人评论,赶快抢个沙发