 腾讯登录
腾讯登录【Nature子刊】开启癌症治疗新篇章!华西药学院秦勇团队揭示生物碱环巴胺的创新合成方法!
| 导读 | 本研究介绍了来自低价和经济的脱氢-表-雄甾酮的藜芦曲明和环巴胺的可扩展合成方法,总产率分别为 11% 和 6.2%。 |
2024年6月22日,四川大学华西药学院秦勇团队在期刊《Nature Communications》上发表了题为“Divergent and gram-scale syntheses of (–)-veratramine and (–)-cyclopamine”的研究论文。藓曲明和环巴胺是异甾体生物碱中最具代表性的两种成员,也是农业和药物化学中有价值的分子。这些化合物的植物提取,受到供应不确定的影响,本研究提供了迄今为止最有效和最多样化的合成方法。

https://www.nature.com/articles/s41467-024-49748-2
研究背景
01
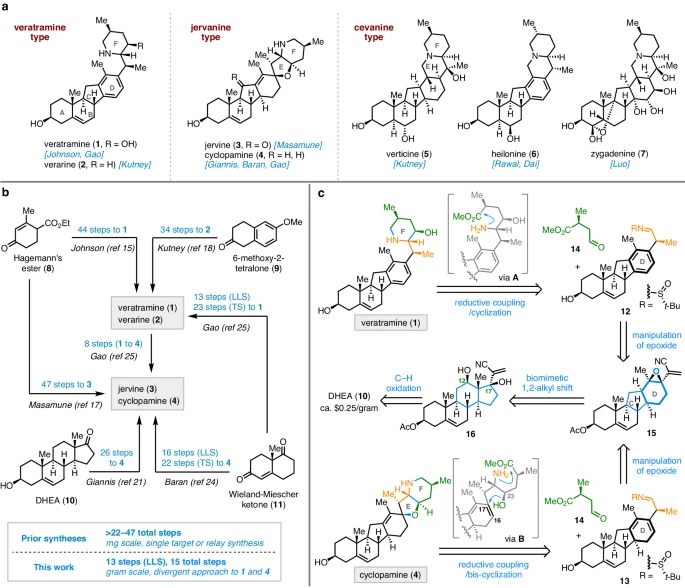
等甾体生物碱(ISA,例如,1-7;无花果。)是来自Veratrum属和Fritillaria属植物的特征化学成分。在结构上,ISA具有共同的C-nor-D-homo类固醇骨架,可分为3种结构类型(即藜芦曲明、杰瓦宁和塞瓦宁)。这些独特的分子表现出各种生物活性,包括显著的镇痛、抗癌、镇咳和杀虫作用。特别是藜芦曲胺类生物碱的代表性成员藜芦曲胺,在中国已被用作商业化的生物农药。此外,杰瓦宁生物碱环巴胺被鉴定为Hedgehog信号通路的有效抑制剂,这促成了一类新型癌症疗法的发展。值得注意的是,patidegib[SGT-610,以前称为saridegib(IPI-926)],目前正处于治疗基底细胞痣综合征(Gorlin 综合征)的III期临床试验中。
背景和研究概要
研究进展
02
ISA的化学和生物学意义,使它们成为合成物质中有吸引力的目标。到目前为止,全球化学家已经合成了7种ISA。具体来说,1967年,Johnson和Masamune开创性地完成了藜芦曲敏的全合成。1968年,Kutney等人发表了一种34步的合成方法。2023年,Baran团队公开了从Wieland-Miescher酮开始的环巴胺的收敛全合成,分16个步骤(最长线性序列,LLS)和22个总步骤。在这项工作之后,Gao及其同事立即完成了13步(LLS,共23步)中,11步的藜芦曲明的合成,以及环巴胺的8步中的继合成。总的来说,所有先前的合成都提供了来自市面上销售的材料:藓芦曲明和杰瓦宁型ISA。其总步骤超过22-47个,以毫克(mg)为单位。尽管取得了这些进展,但对这两种类型的创新合成,仍然非常可取。本研究报告了藜芦胺和环巴胺的发散、高效和克级合成,分 13 步(LLS,共 15 个步骤)与廉价的 DHEA(10 个,约 0.25 美元/克)。
环巴胺的合成,是按照类似的还原偶联/双环化的策略完成。在完成藜芦曲明和环巴胺的合成后,团队进一步研究了含有C17-OH的类固醇二醇的关键仿生重排的通用性,以访问C-nor-D-homo类固醇。该方法与甲硅烷基醚、叔胺、环氧化物、烯酮和叔醇相容。来自不同类固醇骨架的底物也适用于该方案。该方案以良好的效率,提供重排产物(73%产率)和(82%产率)。此外,团队还研究了C17位的不同取代基。结果,乙烯基、氰基和吡啶基,被证明是可行的单元,52-73%的产率形成,已证明了这一点。这种仿生重排能够在简单、温和的反应条件下,产生不同的C-nor-D-homo类固醇。特别是,在此过程中,形成了环氧树脂官能团,可用于各种转化。
本研究对藜芦曲明和环巴胺的逆合成分析如下。在结构上,靶C-去甲-D-同类固醇生物碱,和具有哌啶F环方面是共同的。但是,前者具有断开的E环和芳香族D环,而后者在具有螺四氢呋喃E环方面,有所不同。团队设想在后期通过亚胺和醛的还原偶联/环化序列,组装F环。值得注意的是,通过亚胺和醛之间的还原偶联,可以采用相同的策略,同时建立E/F环,然后,进行中间体的双环化。高级中间体,将通过仿生1,2-烷基,16位位移形成,以构建C-nor-D-homo类固醇框架。最后,可以通过C12氧化制备,以及DHEA的C17添加 。
为了研究DNA损伤反应的代谢特征,团队结合了蛋白质组学和非靶向代谢组学分析,对接受铂类新辅助化疗(NAC)的患者的术后胃癌标本进行了分析。
团队对藜芦曲明和环巴胺的逆合成分析如下图所示。环巴胺的合成,是按照类似的还原偶联/双环化策略完成的。
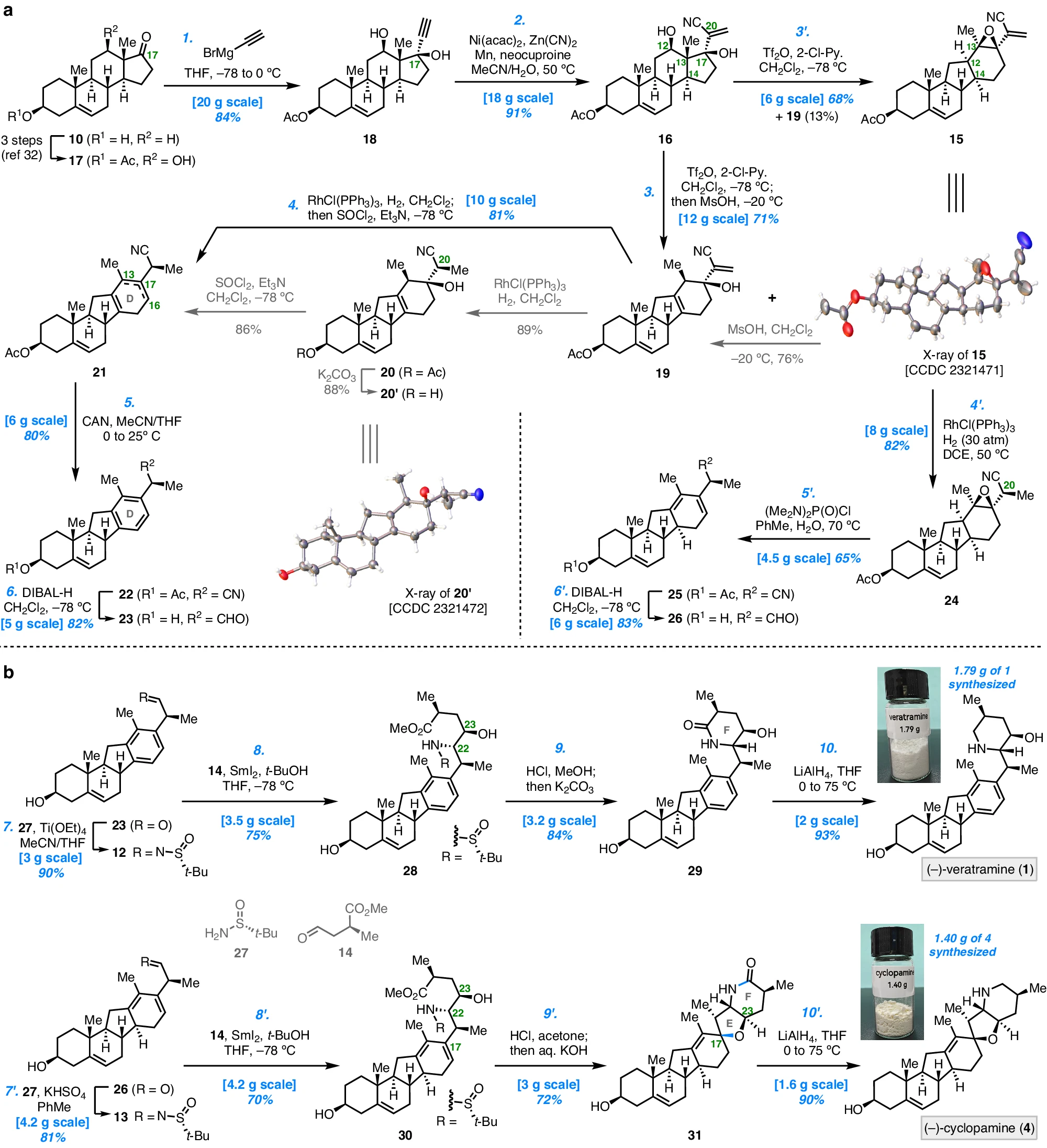
a.制备具有C-nor-D-homo类固醇核心的高级中间体。b.通过还原偶联/环化构建F环,通过还原偶联/双环化构建E/F环。
研究结论
03
迄今为止最有效和最多样化的合成方法,是从市面上销售的低价和经济原料中,提取藜芦胺(13步LLS,11%总产率OY;15个总步骤,6.3%OY)和环巴胺(13步LLS,6.2%OY;15个总步骤,3.6%OY)。本研究通过在克级上执行所有步骤,来证明合成路线的实用性。目前的高效合成,战略性地依赖于1,2-烷基位移形成C-nor-D-homo类固醇核心,环氧部分发散转化为芳烃和共轭二烯,以及立体选择性还原偶联/(双)环化序列来构建(E)/F环体系。值得注意的是,仿生重排方法可以耐受不同的类固醇结构和功能。该方法与氮杂环单元的后期结构相结合,将有助于获得不同的衍生物。此外,虽然目前几乎所有类固醇药物的工业生产,都依赖于半合成,但本研究再次证明了,该方法如何加速复杂和生物活性类固醇的制备。
还没有人评论,赶快抢个沙发